CADD - Breadcrumbs
Welcome! This online resource is designed to provide students, researchers, and professionals with a comprehensive introduction to the fascinating field of Computer-Aided Drug Design (CADD). Here, you will find a wealth of information on the latest computational methods and tools used in drug discovery and development, as well as practical examples and case studies illustrating their application. Whether you are new to CADD or an experienced practitioner, this resource aims to equip you with the knowledge and skills needed to harness the power of computational approaches in the quest for new and improved medicines.
What is Breadcrumbs?
Education stands as a pivotal force in socioeconomic mobility. Yet, generational barriers outside of an individual's influence hinder a person's access to educational opportunities. Breadcrumbs represents OASCI's initiative to fundamentally transform access to educational materials, embodying the spirit of discovery and the sharing of knowledge fragments that have traditionally been accessible only through the goodwill of others. Breadcrumb websites are freely accessible resources, and we encourage contributions, improvements, and adaptations so long as they abide by the terms outlined in the CC BY-NC-SA 4.0 license.
Deploying
We use bump-my-version to release a new version. This will create a git tag that is used by poetry-dynamic-version to generate version strings.
However, we are using Calendar Versioning, meaning we need to specify new versions manually. For example, to bump the version to November 8, 2024, you would run the following command after activating the relevant conda environment.
After releasing a new version, you must push and include all tags.
License
Code contained in this project is released under the MIT License as specified in LICENSE_CODE.
This license grants you the freedom to use, modify, and distribute it as long as you include the original copyright notice contained in LICENSE_CODE and the following notice.
Portions of this code were incorporated with permission from CADD Breadcrumbs by OASCI licensed under the MIT license.
All other data, information, documentation, and associated content provided within this project are released under the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License (CC BY-NC-SA 4.0) as specified in LICENSE_INFO.
Some content was incorporated with permission from CADD Breadcrumbs by OASCI licensed under the CC BY-NC-SA 4.0 license
Web analytics
Why would we want to track website traffic?
An instructor can gain insights into how students engage with online teaching materials by analyzing web analytics. This information is instrumental in assessing the effectiveness of the materials. Web analytics reveal the popularity of specific topics or sections among students, empowering instructors to tailor future lectures or discussions. Analytics also provides valuable data for curriculum development, helping instructors identify trends, strengths, and weaknesses in course materials. Additionally, instructors may leverage web analytics as evidence of their commitment to continuous improvement in teaching methods, which is helpful in discussions related to professional development, promotions, or tenure.
We track website traffic using plausible, which is privacy-friendly, uses no cookies, and is compliant with GDPR, CCPA, and PECR. We also share this website's analytics with you for additional transparency.
Introduction ↵
Computer-aided drug design (CADD) is a rapidly growing field that integrates computational methods and tools to expedite and optimize the drug discovery and development process. By leveraging the power of modern computing, CADD aims to identify novel drug candidates, predict their efficacy and safety, and guide the optimization of lead compounds, ultimately reducing the time and cost associated with bringing new drugs to market.
The origins of CADD can be traced back to the 1960s when the first computational methods for analyzing molecular structures and interactions were developed. However, it was not until the 1980s and 1990s that CADD began to gain significant traction, fueled by advances in computer hardware, software, and the availability of large chemical and biological datasets. Today, CADD has become an integral part of the drug discovery pipeline, with pharmaceutical companies and academic institutions heavily investing in this technology.
The primary goal of CADD is to complement and enhance traditional experimental approaches to drug discovery. By using computational methods, researchers can virtually screen vast libraries of compounds to identify those with the highest potential for binding to a specific biological target, such as a protein or enzyme involved in a disease pathway. This virtual screening process can significantly narrow down the number of compounds that need to be synthesized and tested in the laboratory, saving time and resources.
CADD also plays a crucial role in the optimization of lead compounds. Once a promising drug candidate has been identified, computational methods can be used to predict its absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties. This information can guide the chemical modification of the lead compound to improve its potency, selectivity, and pharmacokinetic profile while minimizing potential side effects.
Another critical application of CADD is in the field of structure-based drug design. By analyzing the three-dimensional structure of a biological target obtained through experimental techniques such as X-ray crystallography or nuclear magnetic resonance (NMR) spectroscopy, researchers can use computational methods to design compounds that precisely fit into the target's binding site. This approach has led to numerous successful drugs, such as HIV protease inhibitors and influenza neuraminidase inhibitors.
The future of CADD is highly promising, driven by continuous advancements in computational power and machine learning algorithms and the increasing availability of big data in the form of genomic, proteomic, and clinical information. Integrating artificial intelligence (AI) and deep learning techniques into CADD is expected to revolutionize the field, enabling the discovery of novel drug targets, the prediction of complex biological interactions, and the generation of new chemical entities with desired properties.
Moreover, the application of CADD is expanding beyond traditional small-molecule drugs to include the design of biologics, such as antibodies and peptides, and the development of personalized medicines tailored to an individual patient's genetic profile. CADD is also vital in repurposing existing drugs for new indications, which can significantly accelerate the drug development process and bring much-needed treatments to patients faster.
In conclusion, computer-aided drug design has emerged as a powerful tool in the quest for new and improved medicines. CADD is transforming the drug discovery landscape by harnessing the power of computational methods and big data. It enables researchers to identify novel drug candidates, optimize their properties, and more efficiently bring innovative treatments to patients. As the field continues to evolve and mature, CADD is poised to play an increasingly critical role in addressing the global health challenges of the 21st century.
History
DRAFT
This page is a work in progress and is subject to change at any moment.
Inception of Computational Techniques
- The application of quantum mechanics to theoretical chemistry in the late 1950s and 1960s, as discussed by Mulliken and Roothaan (1959), highlighted the potential of quantum mechanics for understanding molecular structures and interactions. This marked a critical advancement in the computational approach to drug discovery (Mulliken & Roothaan, 1959).
"The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors" by S. F. Boys and F. Bernardi (1970): This paper presents a new method for computing molecular interactions, focusing on reducing errors in interaction energy calculations. This advancement was crucial for accurately predicting molecular behaviors and interactions in various chemical and biological processes Boys & Bernardi, 1970.
"Equation of state calculations by fast computing machines" by N. Metropolis, A. W. Rosenbluth, M. Rosenbluth, A. H. Teller (1953): This early work described a general method for investigating the properties of substances consisting of interacting molecules, using a modified Monte Carlo integration over configuration space. The method was suitable for fast computing machines of the time, marking a significant step towards modern computational chemistry Metropolis et al., 1953.
"Studies in Molecular Dynamics. I. General Method" by B. Alder and T. Wainwright (1959): This paper outlines a method for calculating the behavior of several hundred interacting classical particles, marking an early step towards the field of molecular dynamics. This technique allowed for the exact computational study of many-body problems, contributing significantly to the understanding of molecular interactions Alder & Wainwright, 1959.
"Liquid Structure and Self‐Diffusion" (1966) by A. Rahman: This paper used the method of molecular dynamics to study the structure of a liquid and its relation to the process of self-diffusion, introducing a precise geometrical procedure to separate particles around a given particle into shells of primary, secondary, etc., neighbors (Rahman, 1966).
"Molecular Dynamics Studies of the Microscopic Properties of Dense Fluids" (1969) by P. L. Fehder: This paper reported molecular dynamics calculations on a two-dimensional system of Lennard-Jones disks, providing equilibrium thermodynamics data for various temperature-density states and examining the presence of large vacancies in the spatial distribution of particles in the system (Fehder, 1969).
- The early 1970s saw the development of versatile and interactive graphics display systems for molecular modeling, as exemplified by the work of Feldman et al. (1973). These systems facilitated the study of macromolecules, marking a significant step forward in the field of CADD (Feldman et al., 1973).
- The late 1970s introduced systems like AIMS (Ames Interactive Molecular modeling System), which utilized 3-D dynamic computer display systems for constructing molecular models and were instrumental in the study of molecular interactions and conformational changes (Coeckelenbergh et al., 1978).
Rise of Structure-Based Drug Design
-
Biophysical Applications of MD: Berendsen's work emphasized the application of MD simulations to complex molecular systems, highlighting its capabilities in predicting the free energy of binding between inhibitors and enzymes. This was instrumental in the application of simulation methods in drug design, demonstrating the potential of MD to contribute significantly to the field (Berendsen, 1987).
-
Chemical and Biomolecular Systems Simulations: Beveridge and Jorgensen's work addressed the simulation of chemical and biomolecular systems, encompassing areas such as free energy simulations and the study of DNA reactions. Their contributions provided a comprehensive overview of the application of computer simulations in understanding biomolecular systems (Beveridge & Jorgensen, 1987).
-
Molecular Dynamics of Proteins: Karplus and colleagues' pioneering efforts in simulating the dynamics of proteins laid the groundwork for the application of MD in studying biomolecules. Their simulations of the bovine pancreatic trypsin inhibitor marked the beginning of molecular dynamics studies of biological macromolecules, setting the stage for future research in protein dynamics and function (Karplus et al., 1987).
-
Constant Pressure and Temperature Simulations: Andersen's development of methods to perform MD simulations under conditions of constant temperature and/or pressure expanded the scope of MD simulations. This allowed for more realistic simulations of biological processes, facilitating the study of biomolecules under varied environmental conditions (Andersen, 1980).
-
Computer-aided Molecular Design: The application of MD simulations in computer-aided molecular design was significantly advanced by Richards, who demonstrated the potential of combining computer graphics techniques with theoretical calculations. This approach was crucial for suggesting molecules with desired specific properties, aiding in the synthetic and chemical manipulation of therapeutic drugs (Richards, 1985).
-
Further developments in the 1970s, including more accurate molecular dynamics simulations, exemplified by Stillinger and Rahman's work on liquid water, underscored the increasing sophistication of computational models in studying molecular properties and interactions (Stillinger & Rahman, 1974).
-
Advances in X-ray Crystallography: The use of high-flux X-ray and neutron solution scattering became instrumental for structural studies of proteins, offering a complement to crystallographic investigations with low-resolution structural methods. This period marked an increase in the quantitative measurements of macromolecular structures and dynamics (Perkins, 1988).
-
Biomolecular Dynamics and Crystallography: Workshops and collaborative efforts underscored the emerging picture of biomolecular dynamics, supported by crystallographic data. This led to a better understanding of enzyme catalysis, nucleic acid functions, and membrane transport, revealing the time dimension in biomolecular interactions (Edholm et al., 1984).
-
Refinement Techniques and Structural Determination: The development of molecular dynamics for the refinement of macromolecular structures demonstrated the feasibility of achieving high-resolution data, critical for understanding biotin-avidin interactions and other complex biomolecular mechanisms (Hendrickson et al., 1989).
-
Emergence of Biological Crystallogenesis: The concept of purity and methodological principles in biological crystallogenesis gained attention, emphasizing the need for a more rational approach to the crystallization of biomacromolecules and their complexes. This was pivotal for the growth of crystals for structural analysis (Giegé & Mikol, 1989).
-
The Protein Data Bank (PDB): The establishment of the PDB as a computer-based archival file for macromolecular structures marked a significant milestone, facilitating the storage and public distribution of atomic coordinates and structural data for a wide array of biomolecules (Bernstein et al., 1977).
-
Fast Energy Estimation and Visualization of Protein-Ligand Interaction: A new computational and graphical method was developed to aid ligand-protein docking studies, capable of estimating non-bonded and electrostatic interaction energy in real-time during interactive docking operations. This method also allowed for the visualization of the local environment inside the binding pocket, significantly aiding the drug design process (Tomioka, Itai, & Iitaka, 1987).
-
Docking Software Development: The late 1980s saw the implementation of docking software packages such as TOM, integrated into FRODO, for studying protein-ligand interactions with interactive energy-minimization procedures. This allowed for the creation of models of protein-ligand complexes, followed by energy minimization treating both ligand and receptor parts as flexible units (Cambillau & Horjales, 1988).
-
Brownian Dynamics Simulation of Protein Association: The application of Brownian Dynamics (BD) to study the diffusive dynamics and interaction of proteins marked an important advancement in understanding protein-protein and protein-ligand interactions. This method assessed the influence of individual charged amino acid residues on the docking process, facilitating the study of electrostatic charge distribution in protein docking (Northrup, Luton, Boles, & Reynolds, 1988).
Evolution of Ligand-Based Drug Design
-
Computer-aided radiopharmaceutical design by Boudreau & Efange (1992) highlights the integration of QSAR with computational methods like quantum mechanics and molecular mechanics, emphasizing the predictive power of QSAR analyses in drug design. This paper underscores the role of computational tools in enhancing the understanding and prediction of drug-radiopharmaceutical interactions (Boudreau & Efange, 1992).
-
Strategies for Indirect Computer-Aided Drug Design by Loew, Villar, & Alkorta (1993) discusses methods used in computer-aided drug design when the structure of the target macromolecule is unknown, emphasizing the indirect characterization of ligands through QSAR and pharmacophore development. This paper provides insight into the challenges and strategies of drug design in the absence of detailed structural information about the target (Loew et al., 1993).
-
Multivariate design and modeling in QSAR by Eriksson & Johansson (1996) explores the crucial steps in developing QSAR models, focusing on the selection of proper data analytical methods, training set design, and validation of QSAR models. This paper highlights the significance of chemometric techniques in QSAR development, relevant to both drug design and environmental sciences (Eriksson & Johansson, 1996).
-
[Development of quantitative structure-activity relationships and computer-aided drug design] by Moriguchi (1994) reviews recent developments in QSAR and computer-aided drug design, including pattern recognition methods for analyzing structure-activity rating data and constructing predictive models for drug design. This paper emphasizes the utility of simple and fast 2D descriptors and molecular mechanical conformational analysis in the QSAR analysis (Moriguchi, 1994).
-
Toward minimalistic modeling of oral drug absorption by Oprea & Gottfries (1999) presents a QSAR model correlating human intestinal absorption and Caco-2 cell permeability data to molecular structures. The model emphasizes a minimalistic approach to predicting oral absorption, integrating hydrophobicity and H-bonding capacity as key descriptors (Oprea & Gottfries, 1999).
Advances in pharmacophore modeling techniques: "Pharmacophore Fingerprinting. 1. Application to QSAR and Focused Library Design" by McGregor & Muskal (1999) introduced a rapid pharmacophore fingerprinting method, showcasing the methodological advancements in pharmacophore modeling during this era (McGregor & Muskal, 1999).
-
Lead generation using pharmacophore mapping and three-dimensional database searching: application to muscarinic M(3) receptor antagonists by Marriott et al. (1999) presents an example of identifying potent novel lead compounds using pharmacophore modeling for muscarinic M(3) receptor antagonists. This study demonstrates the utility of pharmacophore models in 3D database searching and medium-throughput screening to discover compounds with desired biological activity (Marriott et al., 1999).
-
Novel approach to predicting P450-mediated drug metabolism: development of a combined protein and pharmacophore model for CYP2D6 by de Groot et al. (1999) discusses the creation of a combined protein and pharmacophore model for cytochrome P450 2D6 (CYP2D6), integrating pharmacophore modeling, protein modeling, and molecular orbital calculations. This comprehensive approach was used to account for steric, electronic, and chemical stability properties, aligning with experimental data and site-directed mutagenesis results (de Groot et al., 1999).
-
Conformational analysis, pharmacophore identification, and comparative molecular field analysis of ligands for the neuromodulatory sigma 3 receptor by Myers et al. (1994) carried out molecular modeling studies on various ligands showing affinity for the sigma 3 receptor. By employing pharmacophore mapping and comparative molecular field analysis (CoMFA), this study aimed to develop a ligand-binding model for the sigma 3 receptor, demonstrating the application of pharmacophore models in analyzing ligand-receptor interactions (Myers et al., 1994).
Integration of Virtual Screening and High-Throughput Screening (HTS)
-
Integration of Virtual and High-Throughput Screening: This review highlights how high-throughput and virtual screening are complementary components of modern drug discovery, detailing various methods introduced to foster their integration. It emphasizes the potential benefits of a unified approach to biological screening in early-stage drug discovery (Bajorath, 2002).
-
High-throughput and Virtual Screening: Core Lead Discovery Technologies Move Towards Integration: This paper describes the synergies between HTS and virtual screening (VS), discussing developments in VS technology and their potential impact on HTS, including focused screening and data mining (Good, Krystek, & Mason, 2000).
-
Virtual Screening: An Overview: Discusses advances in combinatorial chemistry and HTS, allowing for the synthesis of large numbers of compounds and how virtual screening can reduce a huge virtual library to a manageable size for further evaluation (Walters, Stahl, & Murcko, 1998).
-
Virtual High-Throughput in Silico Screening: This paper covers the application of in silico approaches, such as docking and alignment, to large virtual molecular databases to enrich biologically active compounds, highlighting the cost and time benefits of virtual high-throughput screening (vHTS) (Seifert, Wolf, & Vitt, 2003).
-
Integration of Virtual Screening into the Drug Discovery Process: Reviews advances in virtual screening using docking, predictive ADME methods, and their integration with informatics and computing to enhance drug discovery processes (Chin, Chuaqui, & Singh, 2004).
Advances in Molecular Dynamics Simulations
-
Molecular dynamics simulations of biomolecules by Geisbrecht, B., Gould, S., & Berg, J. (2002). This review highlights the importance of molecular dynamics simulations in understanding the structure and function of biological macromolecules. It discusses the transition from viewing proteins as rigid structures to recognizing their dynamic nature essential for function (Geisbrecht, Gould, & Berg, 2002).
-
Biomolecular simulations: recent developments in force fields, simulations of enzyme catalysis, protein-ligand, protein-protein, and protein-nucleic acid noncovalent interactions by Wang, W., Donini, O., Reyes, C., & Kollman, P. (2001). This paper discusses the cornerstone of computer simulations, the force field, and its applications in understanding enzyme catalysis and macromolecular dynamics and interactions (Wang, Donini, Reyes, & Kollman, 2001).
-
Molecular dynamics simulations of 14 HIV protease mutants in complexes with indinavir by Chen, X., Weber, I., & Harrison, R. (2004). This study utilized molecular dynamics simulations to understand the molecular mechanisms behind HIV drug resistance when interacting with the inhibitor indinavir (Chen, Weber, & Harrison, 2004).
-
Accelerated molecular dynamics: a promising and efficient simulation method for biomolecules by Hamelberg, D., Mongan, J. T., & McCammon, J. A. (2004). The paper presents an accelerated molecular dynamics approach that allows the efficient simulation of biomolecular systems, highlighting its potential in sampling conformational space more efficiently than normal molecular dynamics simulations (Hamelberg, Mongan, & McCammon, 2004).
The Era of Big Data and Machine Learning
-
Deep learning for big data applications in CAD and PLM: This study focuses on the applications of machine learning and deep learning in the manufacturing industry, with a case study on object recognition in heterogeneous formats using deep learning techniques (Dekhtiar et al., 2018).
-
Machine learning on big data: Opportunities and challenges: Discusses the opportunities and challenges of machine learning (ML) in the context of big data, including model scalability and distributed computing (Zhou et al., 2017).
-
Data Mining and Analytics in the Process Industry: Reviews applications of machine learning in the process industry for data mining and analytics, highlighting the role of ML in information extraction, data pattern recognition, and predictions (Ge et al., 2017).
-
Machine Learning With Big Data: Challenges and Approaches: Compiles and summarizes challenges of machine learning with big data, focusing on volume, velocity, variety, or veracity, and discusses emerging ML approaches (L’Heureux et al., 2017).
-
Big data and artificial intelligence (AI) methodologies for computer-aided drug design (CADD): Outlines the development of computational and statistical methods applying big data and AI techniques for CADD, including predictive models for ADMET properties (Lee et al., 2022).
Current Trends and Future Directions
-
Generative AI for Medicinal Chemistry: Generative AI, particularly deep learning generative models, has revolutionized the approach to de novo drug discovery by enabling the design of new medicines through creative computational methods. The application of these AI techniques requires rigorous evaluation to ascertain their real-world utility in drug discovery settings (Walters & Murcko, 2020).
-
Ligand-Based Novel Drug Discovery: Deep learning, through discriminative and generative neural network models, has become crucial for ligand-based novel drug discovery. This area covers virtual screening, neural generative models, and mutation-based structure generation, showcasing the variety and potential of deep learning in generating new molecules with desired properties (Baskin, 2020).
-
Generative Chemistry: Focused on using generative modeling to speed up the drug discovery process, this field explores cutting-edge generative architectures like recurrent neural networks, variational autoencoders, and generative adversarial networks for compound generation. These technologies are key to advancing generative chemistry and, by extension, drug discovery (Bian & Xie, 2020).
-
Deep Reinforcement Learning for Drug Design: This approach integrates generative and predictive models for the design of chemical libraries targeting specific physical and/or biological properties. It exemplifies how AI can generate targeted chemical libraries optimized for desired properties (Popova, Isayev, & Tropsha, 2017).
-
3D Generative Models for Drug Design: Exploring the less-trodden path of 3D molecule generation, DeepLigBuilder offers a novel method for structure-based de novo drug design by generating 3D molecular structures within target binding sites, pushing the frontier in personalized medicine and targeted therapy (Li, Pei, & Lai, 2021).
-
AI in Scaffold-based Design: Generative models are evolving to allow for the incorporation of desired scaffolds directly in the generation process, enabling the creation of novel therapeutic candidates while preserving critical functional groups. This progress is pivotal for designing compounds tailored to specific biological targets, which is a cornerstone of personalized medicine (Joshi et al., 2021).
-
Hybrid Quantum Mechanics/Molecular Mechanics (QM/MM) Simulations for Drug Design: Kulkarni et al. (2021) discuss the application of hybrid QM/MM simulations in structure-based computational methods for therapeutic agent design and discovery, emphasizing their role in high-throughput screening (HTS) and computational chemistry (Kulkarni, Shah, & Vyas, 2021).
-
Scalable Molecular Dynamics with NAMD: Phillips et al. (2020) review NAMD, a program for high-performance molecular dynamics simulations that is versatile for simulations in various thermodynamic ensembles, enabling large-scale simulations on both CPU and GPU architectures (Phillips et al., 2020).
-
Emerging Quantum Computing Algorithms for Quantum Chemistry: Motta and Rice (2021) provide an introduction to emerging algorithms for quantum chemistry simulations on digital quantum computers, focusing on their applications to molecular systems' electronic structure (Motta & Rice, 2021).
-
Exploiting Chemistry for Quantum Information Science: Wasielewski et al. (2020) explore how molecular systems' quantum properties can advance quantum computing, communication, and sensing, highlighting the role of chemistry in the second quantum revolution (Wasielewski et al., 2020).
-
QuantumATK: Integrated Platform for Electronic and Atomic-scale Modelling: Smidstrup et al. (2019) overview QuantumATK, an integrated set of tools for atomic-scale modeling, including electronic-structure calculations and molecular dynamics, supporting various simulation methods (Smidstrup et al., 2019).
-
Full Quantum Eigensolver for Quantum Chemistry Simulations: Wei, Li, and Long (2019) propose a full quantum eigensolver (FQE) algorithm for calculating molecular ground energies and electronic structures on quantum computers, emphasizing faster convergence without a classical optimizer (Wei, Li, & Long, 2019).
-
Recent Advances in First-Principles Based Molecular Dynamics: Mouvet et al. (2022) discuss advances in first-principles molecular dynamics (FPMD) and quantum mechanical-molecular mechanical (QM/MM) extensions for simulating a broad variety of systems (Mouvet et al., 2022).
-
Machine Learning for Molecular Simulation: Noé et al. (2019) review machine learning methods for molecular simulation, focusing on neural networks for predicting quantum-mechanical energies and forces, and their application in molecular dynamics, free energy surfaces, and kinetics (Noé, Tkatchenko, Müller, & Clementi, 2019).
Drug discovery
Drug discovery is critical in the drug development pipeline, laying the foundation for creating safe and effective medicines. This phase involves identifying potential drug candidates that can interact with specific biological targets associated with a disease or condition. The drug discovery process has evolved significantly over the years, with the integration of computational methods playing a crucial role in streamlining and accelerating the identification of promising lead compounds.
Definition and importance
Drug discovery identifies and validates new drug candidates to potentially treat or prevent a specific disease or condition. This phase is the first step in the drug development pipeline and is followed by preclinical studies, clinical trials, and regulatory approval. The success of the entire drug development process heavily relies on the efficiency and effectiveness of the drug discovery phase.
The main objectives of drug discovery are to:
- Identify and validate biological targets associated with a disease or condition
- Discover compounds that can interact with these targets to produce the desired therapeutic effect
- Optimize the properties of these compounds to enhance their efficacy, safety, and drug-like characteristics
Traditional drug discovery
Traditional drug discovery approaches have been the foundation of the pharmaceutical industry for decades. These methods have led to the development of numerous life-saving medications and have significantly improved human health. However, traditional drug discovery is often a time-consuming, expensive, and challenging process with a high attrition rate. Each approach has unique advantages and challenges, and understanding them is crucial for appreciating the evolution and current state of drug discovery.
Serendipitous discovery
Serendipitous discovery, also known as accidental discovery, is a process by which a drug or a potential therapeutic effect is discovered by chance, often while researching something else entirely. Some of the most well-known drugs, such as penicillin, aspirin, and sildenafil, were discovered serendipitously. While serendipitous discoveries have led to significant breakthroughs in medicine, they are, by nature, unpredictable and not a systematic approach to drug discovery. Nevertheless, some methodologies and practices can help foster an environment conducive to serendipitous discoveries:
- Serendipitous discoveries often arise from a researcher's keen observation of unexpected results or phenomena. Curiosity and a willingness to investigate these observations further can lead to identifying novel therapeutic effects.
- Many serendipitous discoveries have emerged from observing unexpected side effects of existing drugs. Investigating these side effects can lead to identifying new therapeutic applications for the drug.
- Natural products, such as plants, fungi, and marine organisms, have been a rich source of serendipitous drug discoveries. Screening these products for biological activity can lead to identifying novel compounds with therapeutic potential.
- Serendipitous discoveries often arise from the cross-pollination of ideas and expertise from different scientific disciplines. Collaborative research and open communication between researchers from various fields can increase the chances of stumbling upon unexpected findings.
While serendipitous discoveries have played a significant role in drug discovery, relying solely on chance is not a sustainable or efficient approach. Modern drug discovery efforts typically involve rational design, high-throughput screening, and other systematic methodologies, with serendipity playing a less prominent role. Nevertheless, fostering an environment that encourages curiosity, collaboration, and exploration can help increase the chances of serendipitous breakthroughs while pursuing more structured approaches to drug discovery.
Phenotypic screening
Phenotypic screening is a drug discovery approach that involves testing compounds in cell-based or animal models to identify those that produce a desired change in the phenotype (observable characteristics) of the disease model without necessarily knowing the specific molecular target of the compound. This approach has the advantage of identifying compounds that modulate disease-relevant pathways and can potentially lead to the discovery of novel drug targets. Here are some common methodologies used in phenotypic screening:
- Pathway-specific reporter assays use genetically engineered cell lines that express a reporter gene (e.g., luciferase or fluorescent protein) under the control of a promoter that is responsive to the activity of a specific signaling pathway. Compounds that modulate the pathway will alter the expression of the reporter gene, providing a measurable readout. Phenotypic profiling involves treating cells with compounds and measuring changes in a wide range of cellular phenotypes using techniques such as gene expression analysis, proteomics, or metabolomics. By comparing the phenotypic profiles of compounds to those of known drugs or genetic perturbations, researchers can gain insights into the compounds' mechanism of action and identify potential drug targets.
- Organoids are three-dimensional, self-organized structures derived from stem cells that recapitulate key features of the organ they represent. Organoid-based screening allows for the testing of compounds in a more physiologically relevant context than traditional 2D cell culture.
- Spheroids are multicellular aggregates that can be generated from tumor cells or other cell types. Spheroid-based screening can be used to identify compounds that inhibit tumor growth, invasion, or other cancer-related phenotypes.
- In vivo screening involves testing compounds in animal models of disease to identify those that produce a desired phenotypic change, such as a reduction in tumor growth, improvement in motor function, or increased survival. In vivo screening provides a more complex and physiologically relevant environment for testing compounds but is also more time-consuming and expensive than in vitro assays.
- Zebrafish have emerged as a powerful model organism for phenotypic screening due to their small size, rapid development, and transparency, which allows for easy visualization of internal organs. Zebrafish can be used to model a wide range of human diseases and can be screened in high-throughput formats to identify compounds that alter disease-related phenotypes.
Phenotypic screening has several advantages over target-based screening, including the ability to identify compounds that act through novel mechanisms and the potential to discover new drug targets. However, phenotypic screening also has limitations, such as the difficulty in determining the specific molecular target of hit compounds and the potential for identifying compounds that produce the desired phenotype through off-target or non-specific effects.
In practice, phenotypic screening is often used with target-based screening and other drug discovery approaches to maximize the chances of identifying new and effective therapies. The choice of screening strategy depends on factors such as the available disease models, the stage of the drug discovery process, and the project's specific goals.
Target-based screening
Target-based screening assays are designed to identify compounds that interact with a specific molecular target, such as a protein or enzyme, that is known to play a role in a particular disease. Several methodologies are used in target-based screening assays, each with advantages and limitations.
- Enzyme inhibition assays measure compounds' ability to inhibit a target enzyme's activity. Compounds are incubated with the enzyme and a substrate, and the enzyme's activity is measured using colorimetric, fluorometric, or radioactive readouts.
- Binding assays assess the direct interaction between a compound and the target protein. Standard techniques include fluorescence polarization, surface plasmon resonance (SPR), and isothermal titration calorimetry (ITC).
- Reporter gene assays use genetically engineered cells that express a reporter gene (e.g., luciferase or green fluorescent protein) under the control of a promoter responsive to the target protein's activity. Compounds that modulate the target's activity will alter the expression of the reporter gene, providing a measurable readout.
- High-content screening (HCS) combines automated microscopy with image analysis software to measure multiple cellular parameters simultaneously. This approach can provide information on a compound's effect on cell morphology, protein localization, and other cellular processes related to the target's function.
- Nuclear magnetic resonance (NMR) spectroscopy can probe the interaction between a compound and a target protein by measuring changes in the protein's chemical shift upon ligand binding. This approach can provide information on the binding site and affinity of the compound.
- X-ray crystallography involves co-crystallizing the target protein with a bound ligand and determining the structure of the complex using X-ray diffraction. This approach can provide detailed information on the binding mode of the compound and guide structure-based drug design efforts.
In practice, target-based screening often involves a combination of these methodologies, with hits identified from one assay being validated and further characterized using additional techniques. The choice of assay depends on factors such as the nature of the target, the available resources, and the stage of the drug discovery process.
It's important to note that while target-based screening has successfully identified new drugs, it also has limitations. For example, compounds that show promising activity in a target-based assay may have a different effect in more complex biological systems, such as cells or animals. Additionally, focusing on a single target may miss compounds that act through novel mechanisms or may not account for the complex interplay between targets in a disease setting.
Role of computer-aided drug design
Despite the successes of traditional drug discovery approaches, the process remains challenging, with high attrition rates and long timelines. The development of a new drug can take more than a decade and cost billions of dollars, and many compounds fail at various stages of the discovery and development process. The pharmaceutical industry has increasingly turned to innovative approaches, such as computer-aided drug design (CADD). CADD techniques offer the potential to streamline the drug discovery process, reduce costs, and increase the likelihood of success by integrating computational methods with traditional approaches. CADD techniques can be broadly categorized into structure-based drug design (SBDD) and ligand-based drug design (LBDD), depending on the availability of structural information about the target or known ligands.
The main advantages of CADD in drug discovery include:
- CADD can rapidly screen vast libraries of compounds and prioritize the most promising candidates for experimental validation, reducing the time and cost associated with traditional screening methods.
- Computational methods can provide valuable insights into the molecular mechanisms underlying drug-target interactions, guiding the optimization of lead compounds.
- CADD enables the rational design of compounds with desired properties, such as enhanced potency, selectivity, and drug-like characteristics.
Overview
The drug discovery phase can be divided into three main stages: target identification, lead identification, and lead optimization.
- Target identification involves identifying and validating biological targets (e.g., proteins, enzymes, receptors, or genes) that play a critical role in the disease or condition of interest. Computational methods, such as bioinformatics and systems biology, can aid in the identification and prioritization of potential targets.
- Once a target has been found, lead identification is used to discover compounds that can interact with the target to produce the desired therapeutic effect. This stage involves screening large libraries of compounds using high-throughput screening (HTS) or virtual screening methods to identify hits that show promising activity against the target.
- The lead optimization stage focuses on improving the properties of the identified hits to enhance their efficacy, selectivity, and drug-like characteristics. This stage involves iterative cycles of chemical modification, biological testing, and computational modeling to guide the optimization process. The ultimate goal is to select one or more optimized lead compounds that can progress to preclinical studies.
In the following sections, we will delve deeper into each stage, exploring the computational methods and strategies employed in modern drug discovery.
-
Chapter 4 of Kumar, T. D. A. (2022). Drug design: A conceptual overview. CRC Press. DOI: 10.1201/9781003298755 ↩
-
Chapter 1 of Rudrapal, M., & Egbuna, C. (Eds.). (2022). Computer aided drug design (CADD): From ligand-based methods to structure-based approaches. Elsevier. ↩
-
Chapter 1 of Strømgaard, K., Krogsgaard-Larsen, P., Madsen, U. (2017). Textbook of drug design and discovery. CRC Press. ↩
Drug development
Following the drug discovery phase, the most promising lead compounds progress through the drug development pipeline. This process involves a series of stages designed to assess potential drug candidates' safety, efficacy, and manufacturability. The drug development process is heavily regulated by agencies such as the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA) to ensure that only safe and effective medicines reach the market.
Preclinical Studies
Preclinical studies are conducted to evaluate the safety and efficacy of lead compounds in non-human models. These studies involve both in vitro (cell-based) and in vivo (animal) experiments to assess the pharmacokinetic and pharmacodynamic properties of the compounds, as well as their potential toxicity.
The main objectives of preclinical studies are to:
- Determine the compounds' absorption, distribution, metabolism, and excretion (ADME) properties.
- Establish the safety profile and identify potential adverse effects.
- Provide evidence of the compound's efficacy in relevant disease or condition animal models.
- Determine the optimal dose and route of administration for further studies.
Investigational New Drug (IND) Application
Before a potential drug candidate can be tested in humans, the sponsor (usually a pharmaceutical company) must file an Investigational New Drug (IND) application with the regulatory agency. The IND application includes data from the preclinical studies and detailed protocols for the proposed clinical trials.
Clinical Trials
Once the IND application is approved, the drug candidate enters the clinical trial stage. Clinical trials are conducted in three main phases.
- Phase 1 These trials involve a small group of healthy volunteers (20-100) and are designed to assess the drug candidate's safety, tolerability, and pharmacokinetics. The main objective is to determine the maximum tolerated dose (MTD) and identify any potential side effects.
- Phase 2 These trials involve a larger group of patients (100-500) with the disease or condition the drug intends to treat. The main objectives are to evaluate the drug candidate's safety and efficacy and to determine the optimal dose for Phase 3 trials.
- Phase 3 These trials involve a large group of patients (1,000-5,000) and are designed to provide definitive evidence of the drug's safety and efficacy. The drug candidate is typically compared to a placebo or the current standard of care treatment. Successful completion of Phase 3 trials is required for regulatory approval.
New Drug Application and Regulatory Approval
Following the successful completion of clinical trials, the sponsor submits a New Drug Application (NDA) to the regulatory agency. The NDA includes all data from the preclinical and clinical studies and information on the manufacturing process and proposed labeling for the drug.
The regulatory agency reviews the NDA to determine whether the drug's benefits outweigh its risks. If the drug is approved, the sponsor can begin manufacturing and marketing the drug.
Post-Market Surveillance
After the drug is marketed, the sponsor must conduct post-market surveillance to monitor the drug's safety and efficacy in the larger patient population. This stage involves collecting and analyzing data on adverse events and other safety concerns, as well as conducting additional studies to evaluate the drug's long-term effects further.
Throughout the drug development process, computational methods can play a crucial role in guiding decision-making and optimizing the chances of success. In the following sections, we will explore how computer-aided drug design (CADD) techniques can be applied at various stages of drug development.
-
Chapter 4 of Rosa, J. M. C. (2023). Pharmaceutical chemistry: Drug design and action. Walter de Gruyter GmbH & Co KG. ↩
-
Chapter 4 of Kumar, T. D. A. (2022). Drug design: A conceptual overview. CRC Press. DOI: 10.1201/9781003298755 ↩
-
Chapter 1 of Strømgaard, K., Krogsgaard-Larsen, P., Madsen, U. (2017). Textbook of drug design and discovery. CRC Press. ↩
Types of CADD
Computer-aided drug design (CADD) is an interdisciplinary field that leverages computational methods to facilitate and accelerate the drug discovery and development process. CADD techniques can be broadly classified into two main categories: structure-based drug design (SBDD) and ligand-based drug design (LBDD). These approaches are often used in combination to provide a more comprehensive understanding of drug-target interactions and to guide the design of new drug candidates.
Structure-Based Drug Design
Structure-based drug design relies on the knowledge of the three-dimensional structure of the target protein or nucleic acid. The main objective of SBDD is to design compounds that can interact with the target's binding site, typically by forming favorable intermolecular interactions such as hydrogen bonds, van der Waals forces, and electrostatic interactions.
The main techniques used in SBDD include:
- Molecular docking involves predicting the orientation and conformation of a ligand within the target's binding site. Docking algorithms evaluate the interactions between the ligand and the target, and rank the ligands based on their predicted binding affinities.
- Molecular dynamics simulations are used to study the dynamic behavior of proteins and protein-ligand complexes. These simulations can provide insights into the stability, flexibility, and conformational changes of the target, as well as the binding mode and residence time of the ligand.
- Structure-based virtual screening involves docking large libraries of compounds into the target's binding site to identify potential hit compounds. The top-ranked compounds are then selected for experimental validation.
- De novo drug design involves designing novel compounds that are complementary to the target's binding site. De novo drug design algorithms generate new molecular structures based on the properties of the binding site, such as its shape, size, and chemical features.
Ligand-Based Drug Design
Ligand-based drug design is used when the three-dimensional structure of the target is not available, but there is information about known ligands that bind to the target. LBDD methods rely on the assumption that structurally similar compounds are likely to have similar biological activities.
The main techniques used in LBDD include:
- Quantitative structure-activity relationship (QSAR) models establish a mathematical relationship between the structural features of a set of compounds and their biological activities. These models can be used to predict the activity of new compounds and guide the design of more potent and selective ligands.
- A pharmacophore is an abstract representation of the essential features of a ligand that are responsible for its biological activity. Pharmacophore models can be used to identify new compounds that share these features and are likely to have similar activity.
- Similarity searching for compounds in chemical databases that are structurally similar to a known active ligand. The underlying assumption is that these similar compounds may also share similar biological activity.
Integrated Approaches and Emerging Techniques
In practice, SBDD and LBDD approaches are often used in combination to provide a more comprehensive understanding of drug-target interactions. For example, pharmacophore models derived from LBDD can be used to guide the docking process in SBDD, while the results of molecular docking can be used to refine QSAR models.
In addition to these established techniques, several emerging approaches in CADD are gaining attention:
- Artificial intelligence (AI) and machine learning (ML) methods, such as deep learning and neural networks, are being increasingly applied to various aspects of drug discovery, including virtual screening, de novo drug design, and ADMET prediction.
- Fragment-based drug design (FBDD) involves identifying small molecular fragments that bind to the target protein and then linking or growing these fragments to create larger, more potent compounds.
- Protein-protein interaction (PPI) inhibitors play a crucial role in many biological processes and are emerging as attractive targets for drug discovery. CADD techniques, such as molecular docking and MD simulations, are being adapted to design small molecules that can disrupt specific PPIs.
In the following chapters, we will delve deeper into each of these CADD techniques, exploring their underlying principles, practical applications, and limitations. We will also discuss how these methods can be integrated into the drug discovery and development pipeline to accelerate the identification of novel therapeutics.
Regulation
Regulatory oversight plays a critical role in ensuring the safety and efficacy of new drugs developed through the drug discovery and development process. As computational methods become increasingly integrated into this process, it is essential to understand the regulatory landscape and how it applies to the use of in silico techniques. This section will provide an overview of the importance of regulatory guidelines for computational methods, specific guidelines for in silico studies, validation requirements for computational models, and the process of moving from computational models to clinical trials.
Importance of regulatory guidelines
Regulatory agencies, such as the United States Food and Drug Administration (FDA) and the European Medicines Agency (EMA), are responsible for overseeing the drug development process and ensuring that only safe and effective medicines reach the market. In recent years, these agencies have recognized the growing importance of computational methods in drug discovery and development and have begun to establish guidelines for their use.
The incorporation of computational methods into regulatory guidelines serves several purposes:
- Ensuring the reliability and reproducibility of computational predictions
- Promoting the use of validated and scientifically sound computational models
- Facilitating the integration of computational data into the drug development process
- Providing a framework for the acceptance of computational evidence in regulatory decision-making
Guidelines for in silico studies
Several regulatory guidelines have been developed to address the use of computational methods in drug discovery and development. These guidelines provide recommendations for the design, execution, and reporting of in silico studies and the validation and documentation requirements for computational models.
One of the most comprehensive guidelines for in silico studies is the International Council for Harmonisation (ICH) M7 guideline, which focuses on the assessment and control of DNA reactive (mutagenic) impurities in pharmaceuticals. This guideline recommends the use of two complementary in silico approaches: (1) expert rule-based and (2) statistical-based, to predict the mutagenic potential of impurities.
Other relevant guidelines include:
- ICH S5 (R3): Detection of Reproductive and Developmental Toxicity for Human Pharmaceuticals
- ICH S1 (R1): Rodent Carcinogenicity Studies for Human Pharmaceuticals
- EMA Guideline on the Reporting of Physiologically Based Pharmacokinetic (PBPK) Modelling and Simulation
These guidelines provide specific recommendations for using computational methods in various aspects of drug discovery and development, such as toxicity prediction, carcinogenicity assessment, and pharmacokinetic modeling.
Validation requirements for computational models
Validation is a critical aspect of ensuring the reliability and acceptability of computational models in the regulatory context. The validation process involves demonstrating that a computational model is fit for its intended purpose and provides accurate and reproducible predictions.
Regulatory agencies typically require the following types of validation for computational models:
- Internal validation: Assessing the model's performance using the data set used to develop the model (e.g., cross-validation).
- External validation: Evaluating the model's performance using an independent data set not used in model development.
- Prospective validation: Testing the model's predictions on new data generated for validation purposes.
In addition to these validation requirements, regulatory agencies also expect detailed documentation of the model development process, including the data sources, algorithms, and assumptions used, as well as any limitations and uncertainties associated with the model.
Moving from Computational Models to Clinical Trials
The ultimate goal of computational methods in drug discovery and development is to identify promising drug candidates that can be advanced to clinical trials. However, transitioning from in silico models to in vitro, in vivo, and human studies requires careful planning and consideration of regulatory requirements.
When incorporating computational findings into Investigational New Drug (IND) applications or New Drug Applications (NDA), it is essential to provide a clear rationale for the use of computational methods, along with supporting evidence from validation studies and relevant literature. Regulatory agencies will consider the strength of the computational evidence in the context of the overall drug development program and may require additional experimental data to support the computational findings.
Challenges in translating computational predictions into clinical insights include:
- Accounting for the complexity and variability of human biology, which computational models may not fully capture.
- Addressing potential discrepancies between computational predictions and experimental results.
- Communicating the limitations and uncertainties of computational methods to regulatory agencies and clinical stakeholders.
Case Studies and Precedents
There have been several notable cases where computational methods have played a significant role in the regulatory approval process. One example is the approval of the anticoagulant drug edoxaban (Savaysa) by the FDA in 2015. The approval of edoxaban was supported by physiologically based pharmacokinetic (PBPK) modeling, which was used to predict the drug's exposure and response in specific patient populations, such as those with renal impairment.
Another example is the use of in silico toxicology models in the safety assessment of new drug candidates. The FDA has recognized the potential of these models to reduce animal testing and streamline the drug development process. In a pilot project, the FDA evaluated the performance of several in silico models in predicting drug-induced liver injury (DILI) and found that some models could accurately identify compounds with a high risk of DILI.
These case studies and precedents demonstrate the growing acceptance of computational methods by regulatory agencies and highlight the potential for these methods to accelerate the drug discovery and development process while ensuring the safety and efficacy of new medicines.
As regulatory agencies continue to refine their guidelines for computational methods and more successful examples of their application emerge, in silico techniques are likely to become an increasingly integral part of the drug discovery and development landscape. Researchers and drug developers who stay informed about the latest regulatory developments and best practices for computational modeling will be well-positioned to leverage these powerful tools to bring new and innovative therapies to patients in need.
Ended: Introduction
Structure based ↵
Structure-based drug design
Structure-based drug design (SBDD) marks a paradigm shift in the world of pharmaceutical development. It moves away from the slow and resource-intensive process of randomly testing vast libraries of molecules. Instead, SBDD leverages the power of computational modeling and detailed knowledge of disease targets. By using the 3D structure of these targets, researchers can virtually design drugs that specifically interact with the disease-causing mechanisms at the molecular level. This targeted approach offers a much faster and more efficient drug discovery process than traditional methods, with the potential for significantly higher success rates. It's akin to having a detailed map guiding you to a successful drug rather than stumbling around in the dark.
Context
Why are we here?
Drug discovery is a complex and often time-consuming process. Traditionally, it involved trial-and-error methods of testing countless molecules to find ones with the desired therapeutic effect. However, the advent of computer-aided drug design (CADD) techniques has revolutionized this field.
Where are we now?
By using the 3D structure of disease-causing molecules, SBDD allows researchers to virtually design drugs that specifically interact and disrupt these mechanisms at the molecular level. This targeted approach offers a much faster, more efficient, and potentially more successful drug discovery route than traditional methods.
Learning objectives
TODO:
Targets ↵
Targets
Biological targets serve as crucial control points at the molecular level. These targets, predominantly proteins like enzymes and receptors, serve as recognition sites for other molecules. This binding event can initiate a domino effect within the cell, influencing fundamental processes like metabolism and intercellular communication. Pharmaceutical research strategically leverages this intricate dance by designing drugs to target these specific structures. These drugs can mimic or inhibit the natural processes the target molecule governs.
-
Chapter 5 of Rosa, J. M. C. (2023). Pharmaceutical chemistry: Drug design and action. Walter de Gruyter GmbH & Co KG. ↩
-
Chapter 2 of Kumar, T. D. A. (2022). Drug design: A conceptual overview. CRC Press. DOI: 10.1201/9781003298755 ↩
-
Chapters 11-14 of Strømgaard, K., Krogsgaard-Larsen, P., Madsen, U. (2017). Textbook of drug design and discovery. CRC Press. ↩
Classes ↵
Target classes
TODO:
DRAFT
This page is a work in progress and is subject to change at any moment.
-
Chapter 5 of Rosa, J. M. C. (2023). Pharmaceutical chemistry: Drug design and action. Walter de Gruyter GmbH & Co KG. ↩
-
Chapter 2 of Kumar, T. D. A. (2022). Drug design: A conceptual overview. CRC Press. DOI: 10.1201/9781003298755 ↩
-
Chapters 11-14 of Strømgaard, K., Krogsgaard-Larsen, P., Madsen, U. (2017). Textbook of drug design and discovery. CRC Press. ↩
Receptors
DRAFT
This page is a work in progress and is subject to change at any moment.
G protein-coupled receptors
Nuclear receptors
-
Chapter 5 of Rosa, J. M. C. (2023). Pharmaceutical chemistry: Drug design and action. Walter de Gruyter GmbH & Co KG. ↩
-
Chapter 2 of Kumar, T. D. A. (2022). Drug design: A conceptual overview. CRC Press. DOI: 10.1201/9781003298755 ↩
-
Chapters 11-14 of Strømgaard, K., Krogsgaard-Larsen, P., Madsen, U. (2017). Textbook of drug design and discovery. CRC Press. ↩
Enzymes ↵
Enzymes
DRAFT
This page is a work in progress and is subject to change at any moment.
TODO:
Kinase
DRAFT
This page is a work in progress and is subject to change at any moment.
TODO:
Protein kinases function as the cell's central command, they exert remarkable control over a multitude of cellular processes through a seemingly simple mechanism: phosphorylation. By transferring phosphate groups derived from adenosine triphosphate (ATP) to specific amino acids on target proteins, protein kinases trigger a cascade of signaling events that precisely regulate cellular destiny.
Biological relevance
Protein kinases occupy a pivotal position in cellular signaling, translating extracellular cues from hormones and growth factors into intricate intracellular responses. These responses encompass a wide range of essential functions:
- Signal Transduction: Kinases act as signal transducers, relaying messages received from external stimuli to initiate critical pathways for cell growth, differentiation, and division.
- Metabolic Control: Protein kinases regulate enzymes involved in energy production and biosynthesis, ensuring efficient cellular metabolism.
- Cell Cycle Progression: These enzymes precisely control the orderly passage of cells through the various stages of the cell cycle, ensuring proper replication and division.
- Cell Fate Determination: Protein kinases influence survival, proliferation, or programmed cell death (apoptosis).
- Gene Expression: Protein kinases can modulate gene expression by regulating transcription factors, dictating which genes are turned on or off in a cell.
Disease development
Given their critical role in cellular control, it's unsurprising that dysregulation of protein kinases can contribute to a multitude of human pathologies:
- Oncogenesis: Aberrant activation of protein kinases is a hallmark of many cancers. These hyperactive kinases promote uncontrolled cell growth and division, a defining characteristic of the malignant state.
- Neurodegenerative Disorders: Dysregulation of protein kinases has been implicated in the pathogenesis of Alzheimer's disease, Parkinson's disease, and Huntington's disease.
- Cardiovascular Diseases: Abnormal protein kinase activity can contribute to the development of heart failure and arrhythmias.
- Chronic Inflammatory Conditions: Inappropriate activation of protein kinases can underlie chronic inflammatory diseases like arthritis.
As a drug target
The pleiotropic effects of protein kinases on cellular processes make them prime targets for drug development. Their attractiveness as therapeutic targets stems from several key advantages:
- Specificity: Drugs can be designed to specifically recognize and inhibit the activity of a particular protein kinase, minimizing off-target effects and improving patient outcomes.
- Diversity: The vast repertoire of protein kinases allows for the targeted modulation of specific signaling pathways implicated in a particular disease.
- Druggability: Many protein kinases possess structural features that facilitate the development of drugs that can effectively bind and modulate their activity.
Additional readings
- Fabbro, D., Cowan‐Jacob, S. W., & Moebitz, H. (2015). Ten things you should know about protein kinases: IUPHAR Review 14. British journal of pharmacology, 172(11), 2675-2700. DOI: 10.1111/bph.13096
Protease
DRAFT
This page is a work in progress and is subject to change at any moment.
TODO:
Proteases, or peptidases, represent a ubiquitous and functionally diverse superfamily of enzymes. Their primary function lies in the hydrolysis of peptide bonds within protein substrates, essentially acting as molecular sculptors that orchestrate a multitude of cellular activities through precise proteolysis. This targeted cleavage regulates critical processes underlying cellular physiology and development.
Biological relevance
Proteases play a pivotal role in maintaining cellular proteostasis. They are instrumental in:
- Digestion: Within the digestive system, a cascade of proteases including pepsin, trypsin, and chymotrypsin facilitates the breakdown of dietary proteins into smaller peptides and amino acids for efficient absorption.
- Protein Turnover: Proteases are essential for the regulated degradation of damaged, misfolded, or short-lived proteins, ensuring the proper functioning of the cellular proteome.
- Signal Transduction: Proteolytic processing of signaling molecules by specific proteases activates or inactivates downstream pathways, influencing cellular responses to diverse stimuli.
- Cell Cycle Regulation: Proteases orchestrate cell division by precisely cleaving regulatory proteins that govern cell cycle progression.
- Extracellular Matrix Remodeling: Proteases are crucial for the dynamic remodeling of the extracellular matrix, a process essential for tissue development, wound healing, and cell migration.
- Immune Response: Proteases participate in the immune system by processing antigen molecules into peptides for presentation to immune cells, facilitating a robust immune response.
Disease development
Tightly regulated proteolytic activity is essential for normal cellular function. However, aberrant protease activity can contribute to the pathogenesis of various diseases:
- Neurodegenerative Disorders: Uncontrolled proteolysis by caspases and other proteases is implicated in the neurodegenerative processes observed in Alzheimer's disease, Parkinson's disease, and Huntington's disease.
- Cancer: Cancer cells often exploit proteases to degrade components of the extracellular matrix, facilitating tumor invasion and metastasis. Additionally, proteases can contribute to tumor growth by activating signaling pathways promoting proliferation and survival.
- Inflammatory Disorders: Excessive proteolytic activity by enzymes like metalloproteases can exacerbate tissue damage and inflammation in diseases like rheumatoid arthritis and inflammatory bowel disease.
- Autoimmune Diseases: Protease malfunction can lead to the inappropriate degradation of healthy tissues, contributing to autoimmune pathologies like rheumatoid arthritis and systemic lupus erythematosus.
As a drug target
The multifaceted roles of proteases in physiology and disease make them attractive targets for drug development. Their suitability stems from several key advantages:
- Specificity: Inhibitor design can target specific proteases based on their active site architecture and substrate recognition, minimizing off-target effects on other proteolytic processes.
- Disease Relevance: Targeting dysregulated proteases offers a strategy to intervene in the pathological processes underlying various diseases.
- Regulation Mechanisms: Many proteases are activated or inhibited by endogenous regulators, providing a framework for the development of drugs that modulate their activity.
Transferase
DRAFT
This page is a work in progress and is subject to change at any moment.
TODO:
Transferases encompass a vast and diverse superfamily of enzymes united by a common task: the transfer of functional groups between donor and acceptor molecules. These "chemical chaperones" orchestrate a multitude of essential cellular processes by facilitating the exchange of moieties like methyl groups, phosphates, glycosyl groups, and amino acids. Due to their functional versatility, transferases play a pivotal role in metabolism, detoxification, and signal transduction, making them crucial for maintaining cellular homeostasis.
Biological relevance
The spectrum of biological functions mediated by transferases is vast:
- Biosynthesis: Transferases are instrumental in the synthesis of essential biomolecules like carbohydrates, lipids, nucleic acids, and amino acids. They facilitate the transfer of functional groups required for building block assembly.
- Metabolism: Transferases are central players in metabolic pathways, catalyzing the transfer of functional groups during energy production, breakdown of nutrients, and the interconversion of metabolites.
- Detoxification: Transferases play a vital role in xenobiotic metabolism, conjugating foreign compounds with functional groups like glutathione, facilitating their elimination from the body.
- Signal Transduction: Certain transferases participate in signal transduction pathways by transferring phosphate groups to signaling molecules, triggering downstream cellular responses.
- Protein Modification: Protein glycosylation, a key post-translational modification, is mediated by transferases, which attach sugar moieties to proteins, influencing their function, stability, and localization.
Disease development
Disruptions in transferase activity can contribute to the pathogenesis of various human diseases:
- Cancer: Altered activity of transferases involved in xenobiotic metabolism can impact the detoxification of carcinogens, potentially promoting cancer development.
- Metabolic Disorders: Defects in transferases involved in carbohydrate or lipid metabolism can lead to imbalances in metabolite levels, contributing to diseases like diabetes and fatty liver disease.
- Neurodegenerative Diseases: Abnormal protein glycosylation mediated by transferases has been implicated in the progression of Alzheimer's disease and Parkinson's disease.
- Autoimmune Diseases: Dysregulation of transferases involved in immune cell activation can contribute to autoimmune pathologies.
As a drug target
Despite their functional heterogeneity, transferases present promising targets for drug development due to several factors:
- Substrate Specificity: Transferases often exhibit high specificity for their substrates and functional groups, allowing for the design of targeted drugs with minimal off-target effects.
- Disease Relevance: Targeting specific transferases involved in disease processes like aberrant xenobiotic metabolism or abnormal protein glycosylation offers a potential therapeutic approach.
- Inhibitor Development: The diverse catalytic mechanisms of transferases provide opportunities for the development of specific inhibitors that can modulate their activity.
Oxidoreductase
DRAFT
This page is a work in progress and is subject to change at any moment.
TODO:
Oxidoreductases, a ubiquitous class of enzymes, orchestrate the intricate dance of electron transfer within living organisms. These "cellular conductors" facilitate the transfer of electrons between electron donors and acceptors, a fundamental process underpinning energy production, metabolism, and the detoxification of reactive molecules. Through their meticulous electron shuttling, oxidoreductases fuel essential cellular functions and maintain a balanced redox state.
Biological relevance
The diverse roles played by oxidoreductases are critical for cellular life:
- Cellular Respiration: The electron transport chain, a cornerstone of cellular respiration, relies heavily on oxidoreductases. These enzymes facilitate the transfer of electrons through a series of protein complexes, ultimately leading to ATP production, the cell's main energy currency.
- Biosynthesis: Oxidoreductases participate in various biosynthetic pathways, providing the necessary electron transfers for the synthesis of essential biomolecules like fatty acids and amino acids.
- Detoxification: These enzymes play a crucial role in detoxifying reactive oxygen species (ROS) generated by cellular metabolism and environmental stressors. Oxidoreductases like superoxide dismutase and catalase work in concert to neutralize these harmful molecules, protecting cells from oxidative damage.
- Signal Transduction: Certain oxidoreductases act as signaling molecules themselves, transferring electrons to trigger specific cellular responses.
- Regulation of Redox Homeostasis: Oxidoreductases maintain a balanced cellular redox state by regulating the ratio of reduced and oxidized molecules. This balance is vital for optimal cellular function and the prevention of oxidative stress.
Disease development
Disruptions in the activity of oxidoreductases can contribute to various human diseases:
- Mitochondrial Diseases: Mutations in genes encoding components of the electron transport chain, often involving oxidoreductases, can lead to mitochondrial dysfunction and a broad spectrum of diseases.
- Neurodegenerative Diseases: Oxidative stress caused by impaired activity of antioxidant enzymes like superoxide dismutase has been implicated in the pathogenesis of Alzheimer's disease and Parkinson's disease.
- Infectious Diseases: Some bacteria and parasites rely on specific oxidoreductases for their survival. Targeting these enzymes can be a potential strategy for developing novel antibiotics.
- Cancer: Dysregulation of redox homeostasis mediated by oxidoreductases can contribute to tumor growth and progression.
As a drug target
Despite their diverse functions, oxidoreductases present potential targets for drug development due to several factors:
- Essential Processes: Targeting oxidoreductases involved in critical processes like the electron transport chain can have a selective effect on pathogenic organisms or dysregulated cellular pathways in cancer.
- Redox Imbalance: Targeting oxidoreductases associated with oxidative stress can offer a therapeutic strategy for diseases like neurodegenerative disorders.
- Inhibitor Design: The well-defined catalytic mechanisms of some oxidoreductases facilitate the development of specific inhibitors that can modulate their activity.
Hydrolase
DRAFT
This page is a work in progress and is subject to change at any moment.
TODO:
Hydrolases, a diverse superfamily of enzymes, wield the power to break down a multitude of chemical bonds using water as a reactant. These "molecular cleavers" play a central role in various cellular processes, from digestion and nutrient breakdown to protein turnover and cellular signaling. Through their precise hydrolysis reactions, hydrolases contribute significantly to maintaining cellular homeostasis and nutrient cycling within organisms.
Biological relevance
The spectrum of biological functions mediated by hydrolases is extensive:
- Digestion: In the digestive system, a cascade of hydrolases like peptidases, lipases, and glycosidases break down complex dietary macromolecules (proteins, fats, and carbohydrates) into smaller components like amino acids, fatty acids, and monosaccharides for efficient absorption.
- Intracellular Digestion: Within cells, hydrolases participate in the degradation of organelles, proteins, and other biomolecules through processes like autophagy (cellular self-cleaning) and lysosomal degradation.
- Metabolism: Hydrolases are crucial for various metabolic pathways, facilitating the breakdown of complex molecules into simpler forms that can be utilized for energy production or building block synthesis.
- Signal Transduction: Certain hydrolases participate in signal transduction by cleaving specific signaling molecules, thereby activating or terminating downstream cellular responses.
- Regulation: Hydrolases can regulate various cellular processes by cleaving regulatory molecules like prohormones, activating them into their functional forms.
Disease development
Dysregulation of hydrolase activity can contribute to the pathogenesis of various human diseases:
- Lysosomal Storage Diseases: Defects in lysosomal enzymes, a specific class of hydrolases, lead to the accumulation of undigested substrates within lysosomes, causing a spectrum of debilitating diseases.
- Digestive Disorders: Insufficient production of digestive enzymes in the pancreas can lead to malabsorption syndromes, where nutrients are not properly broken down and absorbed from food.
- Neurodegenerative Diseases: Altered activity of specific hydrolases has been implicated in the abnormal protein aggregation observed in Alzheimer's disease and Parkinson's disease.
- Autoimmune Diseases: Dysregulation of hydrolases involved in immune cell activation can contribute to autoimmune pathologies.
As a drug target
The diverse functions of hydrolases present both challenges and opportunities for drug development:
- Substrate Specificity: While some hydrolases exhibit high substrate specificity, others have broader activity, making targeted drug design challenging.
- Disease Relevance: Targeting specific hydrolases with known roles in disease processes like lysosomal storage diseases offers a potential therapeutic approach.
- Inhibitor Design: The catalytic mechanisms of hydrolases provide opportunities for the development of inhibitors that can modulate their activity, though achieving high specificity can be difficult.
Lyases
DRAFT
This page is a work in progress and is subject to change at any moment.
TODO:
Lyases, a fascinating class of enzymes, stand apart from their hydrolase counterparts. Unlike hydrolases that utilize water for bond cleavage, lyases achieve the same feat through elimination reactions, forming new double bonds or cyclic structures. These "chemical sculptors" participate in diverse cellular processes, from biosynthesis and energy production to metabolite interconversion and signal transduction. Their unique catalytic mechanisms make them essential players in maintaining cellular homeostasis.
Biological relevance
The spectrum of biological functions mediated by lyases is remarkable:
- Biosynthesis: Lyases play a crucial role in the synthesis of essential biomolecules like amino acids, carbohydrates, and nucleotides. They facilitate the removal of specific functional groups, allowing for the formation of new carbon-carbon bonds or ring structures.
- Metabolism: Lyases participate in various metabolic pathways, enabling the interconversion of metabolites through non-hydrolytic cleavage reactions. This allows for the efficient utilization of substrates and the production of precursors for other metabolic processes.
- Decarboxylation: A prominent function of lyases is the removal of carboxyl groups (CO2) from molecules, a crucial step in energy production pathways like glycolysis and fatty acid metabolism.
- Dehydration: Lyases can remove water molecules from specific substrates, generating new double bonds or cyclic structures. This plays a role in various processes, including prostaglandin synthesis, a pathway involved in inflammation and pain signaling.
- DNA Repair: Certain lyases are involved in DNA repair mechanisms, cleaving damaged DNA strands at specific sites to facilitate their repair and maintain genomic integrity.
Disease development
While the specific roles of lyases in disease development are still being actively investigated, some potential connections are emerging:
- Metabolic Disorders: Mutations in genes encoding specific lyases involved in essential metabolic pathways can lead to imbalances in metabolite levels and contribute to diseases like inherited metabolic disorders.
- Cancer: Dysregulation of lyases involved in the synthesis of essential biomolecules or the breakdown of carcinogens can potentially influence cancer development and progression.
- Bacterial Infections: Some bacteria utilize specific lyases for virulence or toxin production. Targeting these enzymes could be a potential strategy for developing novel antibiotics.
As a drug target
Despite their unique catalytic mechanisms, targeting lyases for drug development presents challenges:
- Diverse Mechanisms: The variety of elimination reactions catalyzed by different lyases makes the design of broad-spectrum inhibitors difficult.
- Essential Functions: Many lyases are involved in critical cellular processes. Targeting them with inhibitors might lead to unwanted side effects.
- Limited Understanding: Compared to other enzyme classes, our understanding of the specific roles of many lyases in disease remains less developed.
Isomerase
DRAFT
This page is a work in progress and is subject to change at any moment.
TODO:
Isomerases, a versatile group of enzymes, orchestrate the intricate rearrangement of atoms within a single molecule. These "molecular sculptors" facilitate the interconversion of isomers, molecules with the same chemical formula but differing in spatial arrangement or bond connectivity. By enabling the conversion between various isomeric forms, isomerases play a pivotal role in metabolism, regulation, and protein folding within living organisms.
Biological relevance
The diverse functions of isomerases are essential for cellular life:
- Metabolism: Isomerases ensure the efficient utilization of metabolites by interconverting various isomers into forms readily used by subsequent metabolic pathways. This is crucial for maintaining energy production, biosynthesis, and the breakdown of nutrients.
- Carbohydrate Metabolism: Isomerases like phosphoglucomutase and triose phosphate isomerase play a vital role in glycolysis, the primary pathway for cellular glucose breakdown, by converting glucose and its intermediates into forms readily used in energy production.
- Amino Acid Metabolism: Isomerases facilitate the interconversion of amino acids into their various stereoisomeric forms, which are essential for protein synthesis and other cellular processes.
- Regulation: Certain isomerases participate in regulatory pathways by converting inactive precursors of regulatory molecules into their active forms, influencing cellular responses.
- Protein Folding: Protein isomerases, a specific class, assist in protein folding by catalyzing the rearrangement of disulfide bonds, ensuring the proper three-dimensional structure and function of newly synthesized proteins.
Disease development
Disruptions in isomerase activity can contribute to the development of various human diseases:
- Metabolic Disorders: Mutations in genes encoding specific isomerases can lead to imbalances in metabolite levels and disrupt essential metabolic pathways. This can contribute to diseases like inherited metabolic disorders, where the body cannot properly utilize certain nutrients due to defective isomerase activity.
- Neurodegenerative Diseases: Altered activity of isomerases involved in protein folding has been implicated in the abnormal protein aggregation observed in diseases like Alzheimer's disease and Parkinson's disease.
- Cancers: Dysregulation of isomerases involved in the metabolism of essential molecules or the activation of signaling pathways can potentially influence cancer development and progression.
As a drug target
The diverse functions of isomerases present both challenges and opportunities for drug development:
- Substrate Specificity: While some isomerases exhibit high substrate specificity, others can act on a broader range of isomers. This complexity can influence the design of targeted drugs.
- Essential Processes: Many isomerases are involved in critical metabolic pathways. Inhibiting them might lead to unwanted side effects.
- Activation/Inhibition Mechanisms: Understanding the mechanisms by which isomerases are activated or inhibited can provide insights for the development of drugs that modulate their activity.
Ligase
DRAFT
This page is a work in progress and is subject to change at any moment.
TODO:
Ligases, also known as synthetases, represent a diverse superfamily of enzymes united by a common task: the formation of new chemical bonds between molecules. These "molecular matchmakers" play a critical role in various cellular processes, from DNA replication and repair to protein synthesis and the assembly of complex molecules. By catalyzing the joining of disparate molecules, ligases orchestrate the construction of essential cellular components and the maintenance of genomic integrity.
Biological relevance
The spectrum of biological functions mediated by ligases is vast:
- DNA Replication and Repair: DNA ligases are indispensable for DNA replication, facilitating the joining of newly synthesized DNA fragments to form a continuous strand. Additionally, they play a vital role in DNA repair processes by sealing nicks or breaks in the DNA backbone.
- Protein Synthesis: Aminoacyl-tRNA synthetases, a specific class of ligases, covalently link amino acids to their corresponding tRNA (transfer RNA) molecules, a crucial step in protein synthesis.
- Metabolic Pathway Interconnection: Ligases can link different metabolic pathways by catalyzing the formation of bonds between specific metabolites, allowing for the efficient utilization of precursors and the production of complex molecules.
- Post-Translational Modifications: Certain ligases participate in post-translational modifications of proteins, attaching functional groups like ubiquitin or small ubiquitin-like modifiers (SUMOs) to regulate protein function, stability, and localization.
- Cell Wall Synthesis: In bacteria and some other organisms, ligases are essential for cell wall construction by catalyzing the formation of glycosidic bonds between sugar molecules.
Disease development
Disruptions in ligase activity can contribute to various human diseases:
- Cancer: Mutations in genes encoding DNA ligases or ligases involved in DNA repair pathways can lead to genomic instability and promote tumorigenesis.
- Neurodegenerative Diseases: Dysregulation of ligases involved in protein ubiquitylation, a key regulatory process, has been implicated in the pathogenesis of neurodegenerative disorders like Alzheimer's disease and Parkinson's disease.
- Autoimmune Diseases: Aberrant protein modifications mediated by ligases can contribute to autoimmune pathologies by altering protein function and triggering inappropriate immune responses.
- Bacterial Infections: Bacterial cell wall synthesis relies on specific ligases. Targeting these enzymes with inhibitors could be a potential strategy for developing novel antibiotics.
As a drug target
The crucial roles played by ligases make them attractive targets for drug development:
- Essential Processes: Targeting ligases involved in critical processes like DNA replication or protein synthesis can have a selective effect on pathogenic organisms or dysregulated pathways in cancer.
- Disease Relevance: Ligases involved in specific disease processes like DNA repair defects or abnormal protein ubiquitylation offer potential therapeutic targets.
- Inhibitor Design: The diverse reaction mechanisms of ligases provide opportunities for the development of specific inhibitors that can modulate their activity.
Ended: Enzymes
Ion channels
DRAFT
This page is a work in progress and is subject to change at any moment.
TODO:
Ligand-gated ion channels
Second-messenger-gated ion channels
Voltage-Gated Ion Channels
-
Chapter 5 of Rosa, J. M. C. (2023). Pharmaceutical chemistry: Drug design and action. Walter de Gruyter GmbH & Co KG. ↩
-
Chapter 2 of Kumar, T. D. A. (2022). Drug design: A conceptual overview. CRC Press. DOI: 10.1201/9781003298755 ↩
-
Chapters 11-14 of Strømgaard, K., Krogsgaard-Larsen, P., Madsen, U. (2017). Textbook of drug design and discovery. CRC Press. ↩
Transporters
TODO:
DRAFT
This page is a work in progress and is subject to change at any moment.
-
Chapter 5 of Rosa, J. M. C. (2023). Pharmaceutical chemistry: Drug design and action. Walter de Gruyter GmbH & Co KG. ↩
-
Chapter 2 of Kumar, T. D. A. (2022). Drug design: A conceptual overview. CRC Press. DOI: 10.1201/9781003298755 ↩
-
Chapters 11-14 of Strømgaard, K., Krogsgaard-Larsen, P., Madsen, U. (2017). Textbook of drug design and discovery. CRC Press. ↩
Ended: Classes
Identification ↵
Target identification
TODO:
-
Chapter 8 of Kumar, T. D. A. (2022). Drug design: A conceptual overview. CRC Press. DOI: 10.1201/9781003298755 ↩
-
Chapter 7 of Renaud, J.-P. (Eds.). (2020). Structural biology in drug discovery: Methods, techniques, and practices. John Wiley & Sons. ↩
-
Chapters 1 and 8 of Strømgaard, K., Krogsgaard-Larsen, P., Madsen, U. (2017). Textbook of drug design and discovery. CRC Press. ↩
Genome-wide association studies
TODO:
Differential gene expression
TODO:
Network analysis
TODO:
Proteomics
TODO:
Ended: Identification
Structure ↵
Target structure
TODO:
-
Chapters 2 and 13 of Kumar, T. D. A. (2022). Drug design: A conceptual overview. CRC Press. DOI: 10.1201/9781003298755 ↩
-
Chapter 7 of Rudrapal, M., & Egbuna, C. (Eds.). (2022). Computer aided drug design (CADD): From ligand-based methods to structure-based approaches. Elsevier. ↩
-
Chapter 15 of Renaud, J.-P. (Eds.). (2020). Structural biology in drug discovery: Methods, techniques, and practices. John Wiley & Sons. ↩
Experimental ↵
Experimental
TODO:
Ended: Experimental
Computational ↵
Computational
Understanding the three-dimensional (3D) structure of proteins is essential for deciphering their functions and interactions. However, determining protein structures experimentally through methods like X-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy can be time-consuming and costly. To overcome these limitations, computational methods have been developed to predict protein structures from their amino acid sequences. We will explore the different approaches used for protein structure prediction, including homology modeling, threading, ab initio methods, and the emerging field of deep learning.
Ended: Computational
Ended: Structure
Binding sites ↵
Binding sites
In the field of drug discovery and design, identifying potential binding sites on target proteins is a crucial step in the development of novel therapeutic agents. These binding sites, often referred to as pockets or cavities, are regions on the protein surface where small molecules, such as drugs or ligands, can interact and form stable complexes. The identification and characterization of these pockets are essential for understanding the function and behavior of proteins, as well as for guiding the design of targeted drug molecules.
Pocket detection ↵
Pocket detection
Pocket discovery, also known as pocket detection or cavity detection, is a computational approach that aims to locate and analyze potential binding sites on proteins. This process involves the use of various algorithms and tools that can efficiently explore the protein surface and identify regions with favorable physicochemical properties for ligand binding. These properties may include the size and shape of the pocket, the presence of specific amino acid residues, and the electrostatic potential of the surrounding environment.
The importance of pocket discovery lies in its ability to narrow down the search space for potential drug binding sites, thereby facilitating the subsequent steps of virtual screening and molecular docking. By focusing on specific regions of the protein that are more likely to accommodate ligands, researchers can save time and computational resources, while increasing the chances of finding promising drug candidates.
Moreover, pocket discovery can provide valuable insights into the structure-function relationships of proteins. By identifying and comparing the binding sites across different proteins, researchers can gain a better understanding of the evolutionary conservation and divergence of these regions, as well as their role in protein-ligand interactions and biological processes.
In the following section, we will explore some of the specific tools and methods commonly used for pocket detection. These tools employ various algorithms and scoring functions to assess the druggability and accessibility of potential binding sites, providing researchers with a powerful toolkit for advancing the drug discovery process.
fpocket
Fpocket 1 uses a sophisticated yet intuitive methodology to identify and analyze potential ligand-binding pockets within protein structures. This open-source software leverages the principles of Voronoi tessellation and alpha spheres, integrated into a comprehensive framework capable of handling large-scale datasets efficiently. At its core, Fpocket employs the concept of alpha spheres. In this geometric notion, a sphere touches four atoms of a protein on its boundary without enclosing any atom within its volume. These spheres are instrumental in delineating the voids and crevices on the protein surface, indicative of potential binding sites.
Voronoi Tessellation and Alpha Sphere Detection
The initial step involves decomposing the protein's 3D space into Voronoi cells using the Qhull algorithm 4. This decomposition facilitates the identification of alpha spheres. Fpocket then filters these spheres based on size criteria to discard those too small (indicative of tight atomic packing) or too large (signifying solvent-exposed regions), focusing on those of intermediate sizes that represent potential binding pockets.
Clustering of Alpha Spheres
Subsequently, Fpocket undertakes a clustering process where alpha spheres in proximity are grouped together, forming clusters that signify potential pockets. This process involves several stages, from initial rough segmentation based on neighbor lists to more refined clustering leveraging multiple linkage criteria, ensuring the comprehensive detection of pocket regions.
Characterization and Ranking of Pockets
Once identified, potential pockets are characterized based on various descriptors, such as size, hydrophobicity, and the types of atoms defining the pocket perimeter. A scoring function, derived using Partial Least Squares (PLS) regression, ranks these pockets based on their potential to bind small, drug-like molecules. This ranking not solely determines drugability but highlights pockets with favorable binding characteristics.
-
Le Guilloux, V., Schmidtke, P., & Tuffery, P. (2009). Fpocket: an open source platform for ligand pocket detection. BMC bioinformatics, 10, 1-11. DOI: 10.1186/1471-2105-10-168 ↩
-
Schmidtke, P., Le Guilloux, V., Maupetit, J., & Tuffery, P. (2010). Fpocket: Online tools for protein ensemble pocket detection and tracking. Nucleic acids research, 38, W582-W589. DOI: 10.1093/nar/gkq383 ↩
-
Kochnev, Y., & Durrant, J. D. (2022). FPocketWeb: Protein pocket hunting in a web browser. Journal of Cheminformatics, 14(1), 58. ↩
-
Barber, C. B., Dobkin, D. P., & Huhdanpaa, H. (1996). The quickhull algorithm for convex hulls. ACM Transactions on Mathematical Software (TOMS), 22(4), 469-483. DOI: 10.1145/235815.235821 ↩
Ended: Pocket detection
Pocket properties
TODO:
Ended: Binding sites
Receptor theories ↵
Receptor theories
TODO:
-
Chapter 2 of Kumar, T. D. A. (2022). Drug design: A conceptual overview. CRC Press. DOI: 10.1201/9781003298755 ↩
-
Chapter 2 of Rudrapal, M., & Egbuna, C. (Eds.). (2022). Computer aided drug design (CADD): From ligand-based methods to structure-based approaches. Elsevier. ↩
Occupancy theory
TODO:
Rate theory
TODO:
Induced-fit theory
TODO:
Macromolecular-perturbation theory
TODO:
Activation-aggregation theory
TODO:
Ended: Receptor theories
Ended: Targets
Binding ↵
Binding
TODO:
Context
Why are we here?
TODO:
Where are we now?
TODO:
Learning objectives
TODO:
Kinetics
At its core, the binding of a protein (\(P\)) and ligand (\(L\)) to form a complex (\(PL\)) is a dynamic process involving repeated association and dissociation events:
The double arrow (\(\rightleftharpoons\)) indicates reversibility: the complex \(PL\) can dissociate back into free protein \(P\) and ligand \(L\).
Rates of reactions
Reaction rates are the speed at which a chemical—or binding—reaction proceeds; in other words, the rate of "conversion" or "binding". This conversion must be a function of concentration, because the frequency of collisions between reactant molecules, which is necessary for a reaction to occur, depends on the concentrations of the reactants. This can be understood through the collision theory of chemical reactions.
According to collision theory, for a reaction to happen, the reactant molecules must:
- Collide with each other;
- Have sufficient energy to overcome the activation energy barrier;
- Have the proper orientation during the collision.
The concentration of reactants directly affects the first factor, the frequency of collisions. Here's why:
- Imagine a fixed volume containing reactant molecules. As the concentration of reactants increases, more molecules are present in the same volume.
- With more molecules in the same space, the average distance between them decreases, making collisions more likely.
- A higher frequency of collisions means that there are more opportunities for reactant molecules to collide with sufficient energy and proper orientation, thus increasing the reaction rate.
Mathematically, the relationship between concentration and collision frequency can be derived using the kinetic theory of gases. The collision frequency (\(Z\)) between two types of molecules (\(A\) and \(B\)) is proportional to the product of their concentrations:
We can define the rate of the forward (binding) reaction as \(k_{on}[P][L]\), where \(k_{on}\) is the association rate constant (units of \(s^{-2}\)) and \([P]\) and \([L]\) are the concentrations of free protein and ligand, respectively. Similarly, the rate of the reverse (dissociation) reaction is \(k_{off}[PL]\), with \(k_{off}\) being the dissociation rate constant (units of \(s^{-1}\)) and \([PL]\) the concentration of the protein-ligand complex.
Equilibrium constants
In binding reactions, there are two main types of equilibrium constants: the association constant (\(K_a\)) and the dissociation constant (\(K_d\)). These constants provide information about the strength and reversibility of the binding interaction between a protein (\(P\)) and a ligand (\(L\)). In both cases, we can we can write a differential equation expressing the rate of change of \([PL]\) as the difference between its formation and breakdown at any point in time.
This is an important equation that allows us to model the kinetics of binding.
If \([PL]\) is changing (i.e., nonzero), this means that our system is not in equilibrium; thus, the forward and reverse conversions become equal giving \(d[PL]/dt = 0\). We can then rearrange our expression and get the following.
where the subscript \(eq\) denotes equilibrium concentrations.
Dissociation
A dissociation constant (\(K_d\)) is a type of equilibrium constant that measures the tendency of a complex to separate into its constituent components. This process is reversible, meaning that the components can also recombine to form the complex. For our protein-ligand complex, \(K_d\) is the ratio to our dissociation (\(k_{off}\)) to our association (\(k_{on}\)) rates. We can rearrange our expression above to solve for \(k_{off} / k_{on}\).
Note
This expression is only valid when we have one-to-one binding. Meaning one ligand binds to one protein. If we had any other stoichiometry such as two ligands binding to one protein, our expression would be invalid.
This shows the important relationship between the kinetic constants \(k_{on}\) and \(k_{off}\) and the equilibrium dissociation constant \(K_d\). A higher \(k_{on}\) or lower \(k_{off}\) will result in a lower \(K_d\), indicating tighter binding at equilibrium. Faster binding or slower unbinding will shift the equilibrium towards the bound complex \(PL\).
A lower Kd value indicates a stronger binding affinity, as it means that a lower concentration of ligand is needed to achieve 50% occupancy of the protein binding sites.
Use \(K_d\) when you want to:
- Compare the binding affinities of different ligands for a given protein;
- Determine the concentration of ligand needed to achieve a desired level of protein binding;
- Estimate the fraction of protein bound to ligand at a given ligand concentration.
The dissociation constant \(K_d\), as derived above, is a key parameter characterizing binding affinity at equilibrium. It has units of concentration (typically M, mM, μM, nM, etc.), and corresponds to the concentration of free ligand at which half the protein is bound. To see this, consider that the fraction of protein bound (\(f_{bound}\)) is given by:
Setting \(f_{bound} = 0.5\) and solving for \([L]\) gives \([L]_{50} = K_d\). Thus, a lower \(K_d\) implies that less free ligand is needed to achieve 50% binding, indicating a higher affinity interaction.
Measuring \(K_d\) experimentally is a key goal in characterizing protein-ligand interactions. One common approach is to perform a titration experiment, measuring some signal that reports on the fraction of protein bound (e.g. fluorescence, radioactivity, etc.) as a function of increasing \([L]\). Fitting the resulting binding curve to the equation above yields \(K_d\). More sophisticated methods like isothermal titration calorimetry (ITC) can provide \(K_d\) as well as enthalpy and entropy of binding from a single experiment by measuring heat evolved. Finally, \(K_d\) can also be obtained from separate measurements of \(k_{on}\) and \(k_{off}\) using techniques like surface plasmon resonance (SPR).
Association
The association constant (\(K_a\)) is the reciprocal of the dissociation constant and is defined as:
\(K_a\) has units of inverse concentration (typically M-1) and represents the tendency of the protein and ligand to form the protein-ligand complex. A higher Ka value indicates a stronger binding affinity.
Use \(K_a\) when you want to:
- Express the binding affinity in terms of complex formation rather than dissociation;
- Compare the binding affinities of different protein-ligand pairs;
- Calculate the Gibbs free energy change (\(\Delta G\)) of the binding reaction using the relationship \(\Delta G = -R T \ln(K_a)\).
Thermodynamics
Protein-ligand binding is a fundamental process that underlies virtually all biological processes, from enzyme catalysis and signal transduction to the action of drugs. Therefore, a deep understanding of the thermodynamics that governs the recognition and association of proteins with their ligands is critical for fields such as biochemistry, cell biology, and especially drug discovery.
Binding free energy
The affinity of a protein-ligand interaction is ultimately determined by the change in free energy upon binding, denoted as \(\Delta G_{bind}\). This is the key thermodynamic quantity that dictates the equilibrium populations of free and bound species. For the general noncovalent binding reaction \(P + L \rightleftharpoons PL\), we can write:
where \(R\) is the universal gas constant, \(T\) is the absolute temperature in Kelvin, and \(K_{a}\) is the association constant at equilibrium.
Derivation
See the literature for a derivation of this expression 4.
A negative \(\Delta G_{bind}\) implies that the binding process is spontaneous and favored, as the products (the bound complex) have lower free energy than the reactants (the unbound molecules). The more negative the \(\Delta G_{bind}\) value, the greater the driving force for binding, and thus the higher the binding affinity between the two molecules. A very negative \(\Delta G_{bind}\) corresponds to a tight or high-affinity binding interaction. Conversely, a positive \(\Delta G_{bind}\) would indicate that the binding is non-spontaneous and unfavorable from a thermodynamic standpoint (although it may still occur under certain kinetic conditions).
Energy partitioning
While \(\Delta G_{bind}\) provides an overall thermodynamic picture, it is often valuable for partition this free energy change into its constituent enthalpic (\(\Delta H\)) and entropic (\(-T \Delta S\)) components:
This partitioning can provide deeper insights into the forces driving or opposing the binding interaction.
Enthalpic contributions
The enthalpic term (\(\Delta H_{bind}\)) accounts for energetic factors like hydrogen bonding, van der Waals interactions, and desolvation effects that stabilize the bound complex. Only a brief overview is provided here; we will go into more detail later.
Hydrogen bonding
Hydrogen bonds are formed when a hydrogen atom bonded to an electronegative atom (such as oxygen or nitrogen) interacts with another electronegative atom. In protein-ligand binding, hydrogen bonds can form between suitable donor and acceptor groups on the ligand and the protein. The strength of a hydrogen bond typically ranges from 2 to 10 kcal/mol, making it one of the strongest non-covalent interactions. The formation of hydrogen bonds between the ligand and the protein can significantly stabilize the complex and contribute favorably to the binding enthalpy. The number, strength, and geometry of hydrogen bonds can vary depending on the specific ligand and protein involved.
van der Waals interactions
Van der Waals interactions are weak, short-range attractive forces that arise from transient dipoles induced by fluctuations in the electron clouds of atoms. These interactions include London dispersion forces and dipole-induced dipole interactions. Although the strength of individual van der Waals interactions is small (typically less than 1 kcal/mol), the cumulative effect of many such interactions can be significant in protein-ligand binding. The shape complementarity between the ligand and the protein's binding site can maximize these interactions, making a favorable contribution to the binding enthalpy.
Electrostatic interactions
Electrostatic interactions involve the attraction or repulsion between charged or partially charged atoms. In proteins, charged amino acid side chains (such as lysine, arginine, aspartate, and glutamate) can interact with charged or polar groups on the ligand. These interactions can include salt bridges (between oppositely charged groups), charge-dipole, and dipole-dipole interactions. The strength of electrostatic interactions depends on the distance between the interacting atoms and the medium's dielectric constant. In an aqueous environment, the dielectric constant is high, which can attenuate the strength of electrostatic interactions. However, in a protein's hydrophobic core, the dielectric constant is lower, making electrostatic interactions more significant. The formation of favorable electrostatic interactions can contribute to the binding enthalpy.
Desolvation
Desolvation refers to removing water molecules from the binding interface of the protein and the ligand. Water molecules form hydrogen bonds with polar and charged groups on the protein and the ligand. When a ligand binds to a protein, some water molecules are displaced, breaking the hydrogen bonds. Breaking hydrogen bonds is an enthalpically unfavorable process, as it requires energy. The magnitude of the desolvation penalty depends on the number and strength of the broken hydrogen bonds. However, releasing water molecules into the bulk solvent can also lead to a favorable entropic contribution, as the water molecules gain translational and rotational freedom.
Conformational changes
Protein-ligand binding can induce conformational changes in the protein, the ligand, or both. These conformational changes can involve breaking or forming intramolecular interactions, such as hydrogen bonds, van der Waals, or electrostatic interactions. The breaking of intramolecular interactions is enthalpically unfavorable, while the formation of new interactions is favorable. The net enthalpic contribution of conformational changes depends on the balance between the broken and formed interactions. Conformational changes can also lead to entropic effects, as the system may lose or gain conformational flexibility.
π-π stacking
In proteins and aromatic groups in ligands, π-π stacking interactions occur between aromatic rings, such as those found in phenylalanine, tyrosine, and tryptophan side chains. These interactions arise from the attraction between the delocalized π electrons of the aromatic rings. The strength ofπ-π stacking interactions depends on the distance and relative orientation of the rings, with a parallel displaced or T-shaped arrangement being the most favorable orientation. The formation of π-π stacking interactions can contribute favorably to the binding enthalpy, with a typical strength of 1-2 kcal/mol per interaction.
Metal coordination
Some proteins, such as zinc, calcium, or magnesium, contain metal ions in their binding sites. Ligands with suitable donor atoms (e.g., oxygen, nitrogen, or sulfur) can coordinate with these metal ions, forming metal-ligand bonds. The strength of metal-ligand bonds depends on the nature of the metal ion and the ligand donor atom, but they can be quite strong (10-30 kcal/mol). The formation of metal-ligand bonds can significantly contribute to the binding enthalpy and provide specificity and orientational constraints on ligand binding.
Entropic contributions
The entropic term (\(-T\Delta S_{bind}\)) captures the opposing effect of reducing configurational freedom upon binding.
Conformational entropy
Conformational entropy refers to the number of accessible conformational states of the protein and the ligand. Upon binding, the protein and the ligand may lose conformational flexibility, leading to a decrease in conformational entropy. This loss of entropy is unfavorable and opposes binding. However, in some cases, binding may stabilize a particular protein or ligand conformation, reducing the entropic penalty. The magnitude of the conformational entropic change depends on the flexibility of the protein and the ligand, as well as the specific conformational constraints imposed by binding.
Desolvation entropy
As mentioned earlier, the desolvation of the binding interface involves the displacement of water molecules from the protein and the ligand surfaces. While breaking water-protein and water-ligand hydrogen bonds is enthalpically unfavorable, releasing these water molecules into the bulk solvent leads to a favorable entropic contribution. The released water molecules gain translational and rotational freedom, increasing the system's overall entropy. The magnitude of the desolvation entropic gain depends on the number of water molecules released and their degree of order in the bound state.
Translational and rotational entropy
Upon binding, the protein and the ligand lose translational and rotational degrees of freedom as they become constrained in the bound complex. This translational and rotational entropy loss opposes binding and is often the most significant entropic penalty. The magnitude of this entropic cost depends on the ligand's size and shape and the protein binding site. Larger and more flexible ligands tend to have a higher entropic penalty upon binding.
Solvent entropy
In addition to desolvating the binding interface, protein-ligand binding can also affect the entropy of the surrounding solvent. If the ligand is hydrophobic, binding to a hydrophobic pocket in the protein can release ordered water molecules from the pocket, resulting in a favorable entropic contribution. On the other hand, if the ligand is charged or polar, its binding may disrupt the local water structure, leading to an unfavorable entropic effect.
Vibrational entropy
Vibrational entropy arises from the vibrational modes of the protein and the ligand. Upon binding, the vibrational modes of the protein and the ligand may be altered, leading to changes in vibrational entropy. However, the contribution of vibrational entropy to the overall binding entropy is usually tiny compared to the other factors.
Conclusions
After discussing the thermodynamic basis of protein-ligand binding, including the key contributions to the binding free energy, enthalpy, and entropy, a natural next step is to consider how we can calculate or estimate these energetic quantities. While experimental techniques can measure overall binding affinities, computational methods are invaluable for dissecting binding thermodynamics in atomic detail.
One of the most widely used computational approaches is molecular mechanics. Molecular mechanics methods use classical physics to model the potential energy of a molecular system as a function of its conformation. The potential energy is calculated using a force field, essentially a set of parameterized equations and atom types that approximate the quantum mechanical energy surface.
With a molecular mechanics force field, one can calculate the enthalpic contributions to binding, such as van der Waals interactions, hydrogen bonding, electrostatics, and so on, based on the molecules' 3D structures. Conformational entropy can also be estimated by sampling an ensemble of low-energy conformations. Molecular mechanics is thus a powerful tool for understanding the driving forces of molecular recognition. Its simplicity makes it indispensable for computationally intensive tasks like molecular dynamics simulations and docking. More broadly, it lays the foundation for various computational approaches in computer-aided drug design.
-
Chapter 2 of Strømgaard, K., Krogsgaard-Larsen, P., Madsen, U. (2017). Textbook of drug design and discovery. CRC Press. ↩
-
Chapters 4 of Anslyn, E. V., & Dougherty, D. A. (2006). Modern physical organic chemistry. University science books. ↩
-
Chapter 9 of Zuckerman, D. M. (2010). Statistical physics of biomolecules: An introduction. CRC press. ↩
-
Gilson, M. K., Given, J. A., Bush, B. L., & McCammon, J. A. (1997). The statistical-thermodynamic basis for computation of binding affinities: a critical review. Biophysical journal, 72(3), 1047-1069. ↩
Enthalpy ↵
Enthalpy
Enthalpy is predominately driven by intermolecular forces or noncovalent interactions that occur between molecules or parts of the same molecule without forming a chemical bond. Although weaker than covalent bonds, these interactions are fundamental to the binding of drugs to their targets and the overall stability of molecular complexes.
-
Chapter 5 of Rosa, J. M. C. (2023). Pharmaceutical Chemistry: Drug Design and Action. Walter de Gruyter GmbH & Co KG. ↩
-
Chapter 5 of Rudrapal, M., & Egbuna, C. (Eds.). (2022). Computer aided drug design (CADD): From ligand-based methods to structure-based approaches. Elsevier. ↩
-
Chapter 2 of Kumar, T. D. A. (2022). Drug Design: A Conceptual Overview. CRC Press. DOI: 10.1201/9781003298755 ↩
-
Chapter 2 of Cooksy, A. (2014). Physical Chemistry: Thermodynamics, statistical mechanics, and kinetics. Pearson. ↩
-
Chapter 2 of Jensen, F. (2017). Introduction to computational chemistry. John wiley & sons. ↩
-
Chapter 2 of Strømgaard, K., Krogsgaard-Larsen, P., Madsen, U. (2017). Textbook of drug design and discovery. CRC Press. ↩
-
Chapter 10 of Cooksy, A. (2014). Physical Chemistry: Quantum chemistry and molecular interactions. Pearson. ↩
-
Chapter 1 of Stone, A. J. (2013). The theory of intermolecular forces. Oxford University Press. ↩
-
Chapters 3 - 4 of Anslyn, E. V., & Dougherty, D. A. (2006). Modern physical organic chemistry. University science books. ↩
Long-range ↵
Long-range interactions
TODO:
Electrostatic ↵
Electrostatic
The electrostatic effects are the simplest to understand in general terms: they arise from the straight-forward classical interaction between the static charge distributions of the two molecules. They are strictly pairwise additive and may be either attractive or repulsive.
Electrostatic forces: interactions between static electric fields
Point charges
These forces arise from the presence of point charges within molecules; for example, ions or partial atomic charges. The interaction between point charges is described by Coulomb's law, which forms the basis for understanding electrostatic forces in molecular systems. Coulomb's law states that the electrostatic force between two point charges (\(q_1\) and \(q_2\)) is directly proportional to the product of their charges and inversely proportional to the square of the distance between them, \(r_{12}\).
The electrostatic potential energy (\(U_{el}\)) is given by:
where \(\varepsilon\) is the dielectric constant of the medium.
What is a dielectric constant?
In this context, the dielectric constant (\(\varepsilon\)) is a measure of a material's ability to reduce the strength of an electric field. In a vacuum, the dielectric constant is equal to 1, while in other media, such as solvents, it is greater than 1. The higher the dielectric constant, the more the medium screens or attenuates the electrostatic interactions between charged particles. When considering electrostatic interactions in molecular simulations, the surrounding environment, such as solvent molecules, can significantly impact the strength of these interactions.
In a vacuum
Imagine two charged particles, let's say two ions, in a vacuum. The electrostatic force between them is given by Coulomb's law, and it depends on their charges and the distance between them. In a vacuum, there is nothing to interfere with this interaction, so the force is relatively strong and long-ranged.
In water
Things are a little different when we consider other media like water.
When an ion is placed in water, the water molecules nearby will reorient themselves due to the ion's electric field. The water molecules are polar, with the oxygen atom having a partial negative charge and the hydrogen atoms having a partial positive charge. If the ion is positively charged (a cation), the oxygen atoms of the water molecules will be attracted to it, pointing their negative ends towards the ion. If the ion is negatively charged (an anion), the hydrogen atoms of the water molecules will be attracted to it, pointing their positive ends towards the ion.
This arrangement of water molecules around the ion creates a hydration shell or solvation shell. The oriented water molecules in this shell generate their own electric field that opposes the electric field of the ion. In other words, the water molecules' dipoles align in a way that partially cancels out the ion's electric field.
Now, let's consider two ions in water. Each ion will have its own hydration shell, with water molecules oriented around it. When the two ions come close to each other, their hydration shells will start to overlap. The water molecules in the overlapping region will be influenced by both ions' electric fields, and their orientation will be a compromise between the two.
This overlapping of hydration shells effectively reduces the direct electrostatic interaction between the two ions. The water molecules in the overlapping region act as a barrier, preventing the ions from feeling the full strength of each other's electric fields. The more water molecules that can fit between the ions (i.e., the greater the distance between the ions), the more effective this screening becomes.
In essence, the water molecules in the hydration shells act as tiny dipoles that can rearrange themselves to counteract the ions' electric fields. This rearrangement of water molecules reduces the strength of the electrostatic interaction between the ions, effectively "screening" or "shielding" the interaction.
It's important to note that this screening effect is not perfect – the ions can still interact with each other through the water molecules, but the interaction is much weaker than it would be in a vacuum. The extent of screening depends on the polarity of the solvent (i.e., its dielectric constant) and the distance between the ions.
Partial atomic charges
In molecules, the distribution of electrons creates regions of positive and negative charge, which can be represented by partial charges assigned to each atom. These partial charges can be determined using quantum mechanical calculations or by fitting to the molecular electrostatic potential (ESP). The ESP is typically sampled at a large number of points around the molecule, and the partial charges are assigned by minimizing the difference between the calculated and fitted potentials.
Multipoles
We discussed the interactions between point charges and the use of Coulomb's law to calculate the forces between them. However, many molecules do not have a net charge, but they still exhibit electric fields due to the uneven distribution of charge within the molecule. These electric fields can be described using the concept of multipole interactions.
Types
We categorize different types of multipoles using the order, \(l\), which is related to the angular dependence of the electric potential and field generated by that charge distribution.
Monopole
A monopole (\(l\) = 1) is a single point charge, such as an atomic ion. The electric field generated by a monopole is spherically symmetric and follows Coulomb's law. The electric potential of a monopole varies as 1/r, where r is the distance from the charge.
Dipole
A dipole (\(l\) = 1) is characterized by a separation \(\left( \overrightarrow{r_i} \right)\) of charges (\(q_i\)) within a molecule.
In a polar covalent bond, such as in the SiO molecule, the electronegative element (O) attracts more electron density from the electropositive element (Si). This results in one end of the bond having a net positive charge, while the other end has a net negative charge. The molecule remains neutral overall, but the uneven charge distribution creates a cylindrically symmetric electric field around the bond, known as a dipole field.
The strength of a dipole is measured by its dipole moment, \(\mu\), which is the product of the charge separation and the distance between the charges. For a system of two point charges \(+q\) and \(-q\) we have
Note that \(\overrightarrow{r_1} + \overrightarrow{r_2}\) can be defined as the Euclidean distance, or magnitude, \(d\). Thus, the dipole moment is
For a dipole, the electric field is strongest along the axis of the dipole and weakens as you move away from this axis. The field lines point from the positive charge to the negative charge. The electric potential of a dipole varies as \(1/r^2\) and has an angular dependence proportional to \(\cos (\theta)\), where \(\theta\) is the angle between the dipole axis and the position vector.
In some molecules, the dipoles of individual bonds may cancel each other out, resulting in a zero net dipole moment. For example, in CO2, the dipoles of the two CO bonds cancel each other, leading to a zero dipole moment. However, the uneven distribution of charges still generates an electric field that is cylindrically symmetric about the bond axis and symmetric under inversion. This type of electric field is characteristic of higher-order multipoles.
Quadrupole
The quadrupole (\(l\) = 2) is the next multipole after the dipole; it is a system of four point charges arranged in a square or tetrahedron, with alternating positive and negative charges. Quadrupole interactions are weaker than dipole interactions but can still play a significant role in intermolecular interactions, especially in the absence of dipole moments.
A quadrupole is a system of four point charges arranged in a square or tetrahedron, with alternating positive and negative charges. The electric field generated by a quadrupole has a more complex angular dependence than that of a dipole. The electric potential of a quadrupole varies as \(1/r^3\) and has an angular dependence proportional to \(3 \cos^2 (\theta) - 1\), where \(\theta\) is the angle between the quadrupole axis and the position vector.
Octopole
The octopole (\(l\) = 3) is an even higher-order multipole, generated by a symmetric arrangement of eight point sources or poles of the electric field. Octopole interactions are generally weaker than quadrupole interactions and are rarely measured in intermolecular effects.
Interactions
All interactions can be expressed as a linear combination of multipole contributions
where \(Y_l^m \left( \theta, \phi \right)\) are the spherical harmonics and the radial dependence is captured in \(U_{l, m} \left( r \right)\).
Monopole-dipole
TODO:
Dipole-dipole
TODO:
Ended: Electrostatic
Induction
Induction is a fundamental concept in understanding long-range intermolecular interactions. It occurs when a molecule's electric field distorts the electronic distribution of a neighboring molecule, inducing a multipole moment in the latter. This induced multipole moment, in turn, interacts with the electric field of the inducing molecule, resulting in an attractive force between the two molecules.
What is polarizability?
In the context of molecular interactions, polarization plays a crucial role in understanding the behavior and properties of molecules and their response to electric fields. When an external electric field is applied to a molecule, it induces a distortion in the molecule's electron distribution, leading to the formation of induced multipole moments. This phenomenon is known as polarization.
Figure 1
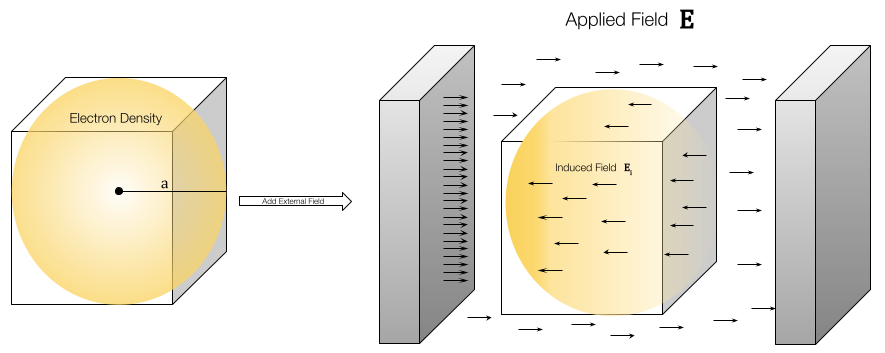
Imagine we have a spherical electron density distribution shown on the left. This negatively charge cloud can be attracted or repelled by shifting its density towards a positive source. We see this effect on the right where we apply a electric field with positive and negative charge on the left and right, respectively.
This is the result of a continuously applied electric field; with induction we instead are polarized by the electric field produced from multipole interactions. Thus, the response can fluctuate based on orientation and other quantum mechanical factors.
Electronic polarization occurs when the electron cloud of an atom within the molecule is displaced relative to the positive nucleus in the presence of an electric field. Polarizability is a measure of a molecule's ability to be polarized in response to an electric field. It is a molecular property that depends on factors such as the electronic structure, molecular geometry, and the strength of the applied field. Molecules with higher polarizability can undergo large shifts of their electron density and exhibit stronger polarization effects.
Induced dipole
Polarization has various effects on the behavior and properties of molecules. One important effect is the induction of dipole moments in neighboring molecules. When a molecule with a permanent dipole moment or an induced dipole moment interacts with a nearby molecule, it can induce a dipole moment in the latter. This induced dipole moment, in turn, interacts with the electric field of the inducing molecule, resulting in an attractive force between the two molecules. This interaction is known as the dipole-induced dipole interaction and contributes to the overall intermolecular forces.
Warning
You can induce high-order multipoles as well, but we will just consider dipoles here.
The induced dipole moment is defined as
where \(\alpha\) is the polarizability of the molecule and \(\mathcal{E}\) is external electric field.
TODO:
TODO:
Dispersion
Dispersion is a type of intermolecular attraction that exists between all atoms and molecules, even those that lack permanent dipole moments or the ability to form hydrogen bonds. This force plays a crucial role in non-covalent binding and becomes the dominant attractive force for molecules containing more than five atoms.
Quantum mechanical origin
The dispersion force originates from the quantum mechanical nature of electrons. In atoms and molecules, electrons are constantly in motion, and their positions are described by wavefunctions. These fluctuations in the electron distribution can create instantaneous dipoles, even in non-polar molecules. An instantaneous dipole occurs when the electron distribution momentarily becomes asymmetric, with more electrons on one side of the molecule than the other. Although the average dipole moment of a non-polar molecule is zero, these instantaneous dipoles can still interact with neighboring molecules.
When an instantaneous dipole forms in one molecule, it can induce a dipole in a nearby molecule through electrostatic interactions. The induced dipole is aligned in such a way that it is attracted to the original instantaneous dipole. This attraction between the instantaneous dipole and the induced dipole gives rise to the dispersion force. The interaction between the molecules leads to a correlation between the motion of their electrons, resulting in a lower overall energy state.
Effect on non-covalent binding
Dispersion forces contribute to the overall stability of non-covalent binding between molecules. In the absence of other strong intermolecular forces, such as dipole-dipole interactions or hydrogen bonding, dispersion forces can be the primary source of attraction.
The strength of the dispersion force depends on several factors, including the size and polarizability of the molecules involved. Larger molecules and those with more easily polarizable electron clouds tend to have stronger dispersion forces. This is because larger molecules have more electrons that can participate in the formation of instantaneous dipoles, and more polarizable electron clouds are more easily distorted by nearby instantaneous dipoles.
As the distance between molecules increases, the magnitude of the dispersion force decreases. At very large separations, the finite speed of light becomes a factor in the interaction between molecules. The correlation between the fluctuations in the electron distributions of the two molecules becomes less effective because the information about a fluctuation in one molecule takes time to reach the other molecule. By the time the second molecule responds and the information about its response reaches the first molecule, the electrons have already moved, and the fluctuations are no longer in phase. This retardation effect leads to a weaker dispersion force at very large separations.
Resonance
TODO:
Ended: Long-range
Short-range interactions
TODO:
Specialized ↵
Specialized
TODO:
Hydrogen bonds
Categorizing and defining hydrogen bonds as a non-covalent interaction has been rife with controversy. IUPAC currently defines a hydrogen bond as 1
The hydrogen bond is an attractive interaction between a hydrogen atom from a molecule or a molecular fragment X—H in which X is more electronegative than H, and an atom or a group of atoms in the same or a different molecule, in which there is evidence of bond formation.
A typical hydrogen bond may be depicted as X—H⋅⋅⋅Y—Z, where the three dots denote the bond. X—H represents the hydrogen bond donor. The acceptor may be an atom or an anion Y, or a fragment or a molecule Y—Z, where Y is bonded to Z. In some cases, X and Y are the same. In more specific cases, X and Y are the same and X—H and Y—H distances are the same as well leading to symmetric hydrogen bonds. In any event, the acceptor is an electron rich region such as, but not limited to, a lone pair of Y or π-bonded pair of Y—Z.
IUPAC provides criteria as evidence or characteristics of a hydrogen bond.
The evidence for hydrogen bond formation may be experimental or theoretical, or ideally, a combination of both. Some criteria useful as evidence and some typical characteristics for hydrogen bonding, not necessarily exclusive, are listed below, numbered E# and C#, respectively. The greater the number of criteria satisfied, the more reliable is the characterization as a hydrogen bond.
(E1). The forces involved in the formation of a hydrogen bond include those of an electrostatic origin, those arising from charge transfer between the donor and acceptor leading to partial covalent bond formation between H and Y, and those originating from dispersion.
(E2). The atoms X and H are covalently bonded to one another and the X—H bond is polarized, the H⋅⋅⋅Y bond strength increasing with the increase in electronegativity of X.
(E3). The X—H⋅⋅⋅Y angle is usually linear (180º) and the closer the angle is to 180º, the stronger is the hydrogen bond and the shorter is the H⋅⋅⋅Y distance.
(E4). The length of the X—H bond usually increases on hydrogen bond formation leading to a red shift in the infrared X—H stretching frequency and an increase in the infrared absorption cross-section for the X—H stretching vibration. The greater the lengthening of the X—H bond in X—H⋅⋅⋅Y, the stronger is the H⋅⋅⋅Y bond. Simultaneously, new vibrational modes associated with the formation of the H⋅⋅⋅Y bond are generated.
(E5). The X—H⋅⋅⋅Y—Z hydrogen bond leads to characteristic NMR signatures that typically include pronounced proton deshielding for H in X—H, through hydrogen bond spin—spin couplings between X and Y, and nuclear Overhauser enhancements.
(E6). The Gibbs energy of formation for the hydrogen bond should be greater than the thermal energy of the system for the hydrogen bond to be detected experimentally.(C1). The pKa of X—H and pKb of Y—Z in a given solvent correlate strongly with the energy of the hydrogen bond formed between them.
(C2). Hydrogen bonds are involved in proton-transfer reactions (X—H⋅⋅⋅Y → X⋅⋅⋅H—Y) and may be considered the partially activated precursors to such reactions.
(C3). Networks of hydrogen bonds can show the phenomenon of co-operativity, leading to deviations from pair-wise additivity in hydrogen bond properties.
(C4). Hydrogen bonds show directional preferences and influence packing modes in crystal structures.
(C5). Estimates of charge transfer in hydrogen bonds show that the interaction energy correlates well with the extent of charge transfer between the donor and the acceptor.
(C6). Analysis of the electron density topology of hydrogen-bonded systems usually shows a bond path connecting H and Y and a (3,—1) bond critical point between H and Y.
TODO:
-
Arunan, E., Desiraju, G. R., Klein, R. A., Sadlej, J., Scheiner, S., Alkorta, I., ... & Nesbitt, D. J. (2011). Definition of the hydrogen bond (IUPAC Recommendations 2011). Pure and applied chemistry, 83(8), 1637-1641. DOI: 10.1351/PAC-REC-10-01-02 ↩
-
Pimentel, G. C., & McClellan, A. L. (1971). Hydrogen bonding. Annual Review of Physical Chemistry, 22(1), 347-385. DOI: 10.1146/annurev.pc.22.100171.002023 ↩
-
Chapter 8 of Stone, A. J. (2013). The theory of intermolecular forces. Oxford University Press. ↩
-
Chapter 2 of Jensen, F. (2017). Introduction to computational chemistry. John wiley & sons. ↩
-
Chapter 10 of Cooksy, A. (2014). Physical Chemistry: Quantum chemistry and molecular interactions. Pearson. ↩
-
Chapters 3 - 4 of Anslyn, E. V., & Dougherty, D. A. (2006). Modern physical organic chemistry. University science books. ↩
Hydrophobicity
Hydrophobic and hydrophilic interactions play a crucial role in the behavior of molecules in aqueous environments, particularly in biological systems. These interactions are governed by a complex interplay of electrostatic forces, induction forces, and entropy, which collectively determine water molecules' solubility and aggregation.
Hydrophilic
Hydrophilic interactions occur between polar or charged molecules with a high affinity for water. Electrostatic forces primarily drive these interactions, including charge-charge interactions (ion-ion), charge-dipole interactions (ion-dipole), and dipole-dipole interactions. In an aqueous environment, polar or charged molecules interact favorably with water by forming hydrogen bonds. Water molecules orient themselves around the polar or charged solute, forming a hydration shell stabilizing the solute in solution. Induction forces, known as polarization forces, contribute to hydrophilic interactions. These forces arise when a polar or charged molecule induces a dipole in a nearby molecule, leading to an attractive interaction. The strength of hydrophilic interactions depends on the magnitude of the electrostatic forces involved. Molecules with higher charge densities or stronger dipole moments exhibit stronger hydrophilic interactions and higher solubility in water.
Hydrophobic
Conversely, hydrophobic interactions occur between nonpolar molecules or regions of molecules that have a low affinity for water. These interactions are driven by water molecules' tendency to minimize their contact with nonpolar solutes, leading to the aggregation of hydrophobic molecules or regions. The primary forces contributing to hydrophobic interactions are dispersion forces, also known as London dispersion forces or van der Waals forces. Dispersion forces arise from temporary fluctuations in the electron density of atoms or molecules, forming transient dipoles. In an aqueous environment, water molecules form a highly ordered network of hydrogen bonds. When a nonpolar solute is introduced, it disrupts this network, as water molecules cannot form hydrogen bonds with the solute. To minimize this disruption, water molecules rearrange themselves around the nonpolar solute, forming a hydrophobic hydration shell. This rearrangement reduces the system's entropy, as the water molecules become more ordered. To counteract this decrease in entropy, nonpolar solutes tend to aggregate, minimizing their surface area exposed to water and reducing the overall entropy loss.
Entropic effects play a significant role in both hydrophobic and hydrophilic interactions. Hydrophobic interactions are largely driven by the entropic gain associated with minimizing the hydrophobic surface area exposed to water, known as the hydrophobic effect. The aggregation of hydrophobic molecules or regions allows water molecules to maintain their hydrogen bonding network more effectively, increasing the system's overall entropy. This entropic driving force is the primary reason for the association of nonpolar substances in aqueous environments. The hydrophobic effect is temperature-dependent and becomes stronger at higher temperatures due to the increased significance of the entropic contribution to free energy.
In contrast, hydrophilic interactions are less influenced by entropic effects compared to hydrophobic interactions. The formation of a hydration shell around a hydrophilic solute does not necessarily lead to a significant decrease in entropy, as the water molecules can still maintain a relatively high degree of hydrogen bonding. In some cases, the introduction of a hydrophilic solute can even lead to an increase in entropy, as the solute disrupts the local structure of water and allows for more dynamic hydrogen bonding interactions. However, the entropy changes associated with hydrophilic interactions are generally smaller in magnitude compared to those involved in hydrophobic interactions.
Balancing interactions
Many molecules, particularly biomolecules such as proteins and lipids, possess both hydrophobic and hydrophilic regions. The balance between these regions determines the overall solubility and behavior of the molecule in an aqueous environment. Amphiphilic molecules, such as phospholipids and detergents, have a hydrophobic tail and a hydrophilic head. In aqueous solutions, these molecules self-assemble into structures like micelles or bilayers, with the hydrophobic tails aggregating to minimize their contact with water while the hydrophilic heads interact favorably with the aqueous environment. In proteins, the interplay between hydrophobic and hydrophilic interactions is crucial for their folding and stability. The hydrophobic effect drives the burial of nonpolar amino acid residues in the protein core, while hydrophilic residues tend to be exposed on the surface, interacting with the aqueous surroundings. The balance between these interactions, along with other factors such as hydrogen bonding and van der Waals forces, determines the three-dimensional structure and function of proteins.
Salt bridges
A salt bridge is a type of non-covalent interaction that occurs between two oppositely charged functional groups, typically between the negatively charged carboxylate group (COO⁻) of an acidic amino acid (such as aspartic acid or glutamic acid) and the positively charged amino group (NH3⁺) of a basic amino acid (such as lysine or arginine).
Figure 1
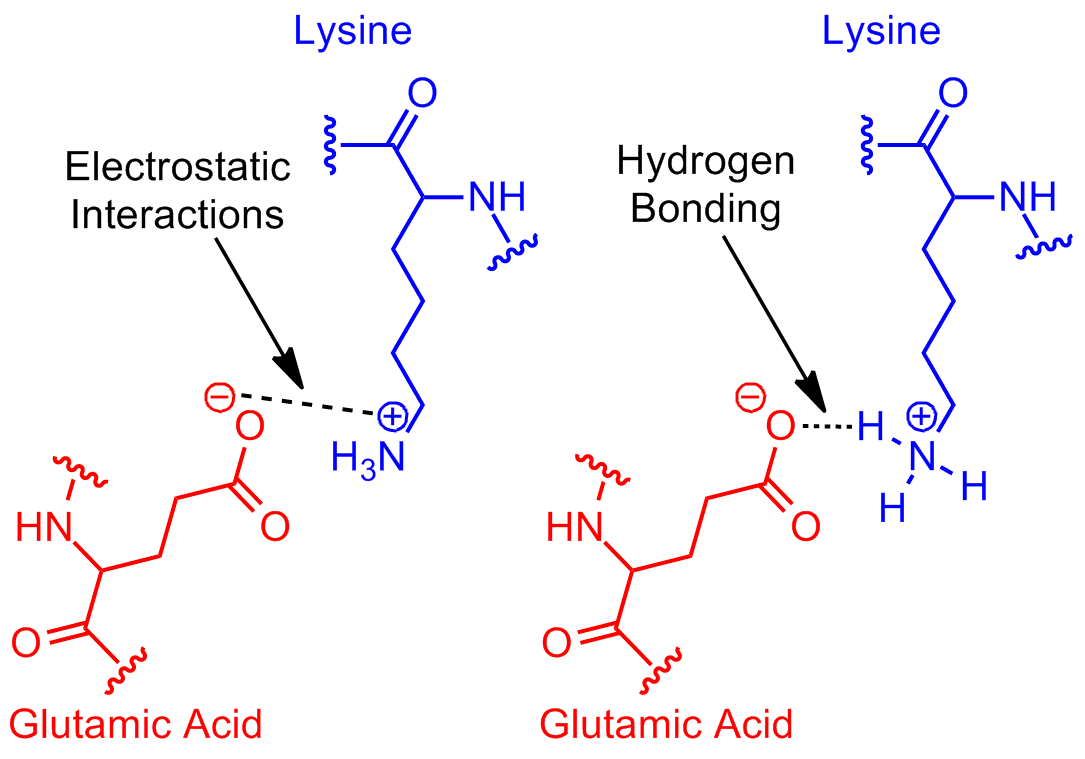
Example of salt bridge between amino acids glutamic acid and lysine demonstrating electrostatic interaction and hydrogen bonding.
A salt bridge is a combination of intermolecular interactions:
- Hydrogen bonding: The hydrogen atom attached to the positively charged amino group (NH3⁺) can form a hydrogen bond with one of the oxygen atoms of the negatively charged carboxylate group (COO⁻). Hydrogen bonds are relatively strong non-covalent interactions that occur when a hydrogen atom bonded to an electronegative atom (such as nitrogen or oxygen) is attracted to another electronegative atom.
- Electrostatic interactions: The opposite charges of the carboxylate group (COO⁻) and the amino group (NH3⁺) attract each other through electrostatic interactions, also known as ionic bonds. These interactions are based on the attraction between oppositely charged ions and contribute to the stability of the salt bridge.
The combination of hydrogen bonding and electrostatic interactions in a salt bridge provides stability to the local structure of a protein or other biomolecule. Salt bridges can be important for:
- Maintaining the tertiary structure of proteins by stabilizing the folded conformation.
- Facilitating protein-protein interactions by providing a favorable interface between two proteins.
- Contributing to the stability of protein complexes, such as enzyme-substrate complexes or receptor-ligand complexes.
- Participating in catalytic mechanisms of enzymes by stabilizing transition states or reaction intermediates.
It's important to note that the strength and stability of salt bridges can be influenced by factors such as the pH and ionic strength of the surrounding environment. Changes in these conditions can affect the protonation state of the acidic and basic amino acids involved in the salt bridge, potentially disrupting the interaction.
π-interactions
π-interactions are types of non-covalent interactions that involve the attractive forces between ions or molecules and the electron-rich π-systems of aromatic rings.
TODO:
Credit: Dr. S. Immel
π-π
Cation-π
Ended: Specialized
Ended: Enthalpy
Entropy
TODO:
-
Chapter 2 of Cooksy, A. (2014). Physical Chemistry: Thermodynamics, statistical mechanics, and kinetics. Pearson. ↩
-
Chapter 6 of Phillips, R., Kondev, J., Theriot, J., & Garcia, H. (2012). Physical biology of the cell. Garland Science. ↩
-
Chapter 3 of Zuckerman, D. M. (2010). Statistical physics of biomolecules: An introduction. CRC press. ↩
Ended: Binding
Docking ↵
Docking
Protein-ligand docking is a computational method that predicts the preferred orientation of a ligand when bound to a protein receptor, forming a stable complex. This technique plays a crucial role in the field of drug discovery and design, as it allows researchers to investigate the interactions between potential drug candidates and their target proteins. By understanding these interactions at the molecular level, scientists can rationalize the design of new drugs, optimize existing compounds, and predict their binding affinities and biological activities.
-
Yang, C., Chen, E. A., & Zhang, Y. (2022). Protein–ligand docking in the machine-learning era. Molecules, 27(14), 4568. DOI: 10.3390/molecules27144568 ↩
-
Paggi, J. M., Pandit, A., & Dror, R. O. (2024). The art and science of molecular docking. Annual Review of Biochemistry, 93. DOI: 10.1146/annurev-biochem-030222-120000 ↩
Preparation ↵
Preparation
Before performing molecular docking simulations, it is crucial to properly prepare both the protein target and the ligands of interest. This preparation step ensures that the molecules are in a suitable format and have the necessary characteristics for accurate and reliable docking results. By carefully preparing the protein and ligands, researchers can enhance the quality and reproducibility of their docking experiments, ultimately leading to more meaningful insights into the binding interactions between these molecules.
Protein
TODO:
Conformation
When selecting a protein structure for molecular docking, the choice between an apo (unbound) or holo (bound) structure is an important consideration. The conformation of the protein can significantly influence the docking results and the predicted binding modes of the ligands.
Ligand-bound structures usually outperform ligand-free structures, as the geometries of the binding pocket are better defined in the bound state than in the unbound state. When a ligand is bound to the protein, it induces conformational changes in the binding site, optimizing the pocket for favorable interactions. These induced-fit effects are captured in the holo structure, providing a more realistic template for docking simulations. In contrast, apo structures may have a less defined or altered binding pocket geometry, as the absence of a ligand can allow for more flexibility or different conformational states.
If a holo structure is not available, computational tools such as SphGen, SiteMap, fpocket, and FTMap can be used to identify potential ligand binding sites. These tools employ various algorithms to predict and characterize binding pockets based on the protein's surface properties, such as shape, electrostatic potential, and hydrophobicity.
However, there are situations where apo structures can still be valuable. For example, if the goal is to identify novel binding sites or explore the protein's conformational flexibility, using an apo structure may be advantageous. Additionally, if the available holo structures have ligands that are significantly different from the ligands of interest, using an apo structure might be more appropriate to avoid bias towards a specific ligand class.
It is also important to consider the quality and resolution of the protein structure. High-resolution structures (e.g., those determined by X-ray crystallography) are preferred for docking studies, as they provide more accurate atomic coordinates. However, if high-resolution structures are not available, homology models or structures derived from other experimental techniques (e.g., cryo-electron microscopy) can be used, albeit with caution.
Mutations
It is crucial to use the wild-type protein structure rather than mutant structures.
However, if a wild-type protein is not available the next best option is to use the minimally mutated structure. Mutant proteins are often used in structural studies due to their enhanced stability, improved crystallization properties, or other biochemical reasons. However, these mutations can alter the protein's structure and function, particularly if they are located within or near the ligand binding site.
To ensure the accuracy and biological relevance of the docking results, it is essential to revert any mutations present in the protein structure back to the wild-type sequence. This process involves carefully examining the protein structure and identifying the mutated residues. Once identified, the mutated residues should be computationally replaced with the corresponding wild-type residues.
It is important to consider the potential structural consequences of reverting the mutations. Mutations can cause local conformational changes in the protein structure, which may affect the shape and properties of the ligand binding site. Therefore, after reverting the mutations, it is advisable to perform energy minimization or structural refinement to allow the protein to adjust to the wild-type sequence and adopt a more natural conformation.
In some cases, the structural impact of reverting mutations may be minimal, especially if the mutations are located far from the ligand binding site. However, if the mutations are within or in close proximity to the binding site, their reversion can significantly influence the docking results. Neglecting to revert such mutations may lead to inaccurate or misleading predictions of ligand binding modes and affinities.
Missing atoms
It is essential to address any missing atoms, side chains, or loops in the experimental protein structures before performing protein-ligand docking. Incomplete protein structures can arise due to limitations in crystallographic resolution, disorder, or experimental challenges.
When preparing the protein structure for docking, special attention should be given to missing side chains and loops that are in close proximity to the binding site. These missing elements can have a significant impact on the shape, size, and chemical properties of the binding pocket, ultimately affecting the accuracy of the docking results.
To address missing side chains, computational tools can be employed to model and reconstruct them based on the available backbone information and known amino acid sequences. This process involves predicting the most likely conformations of the missing side chains while considering their interactions with the surrounding residues and the overall protein structure.
Similarly, missing loops near the binding site should be carefully modeled and reconstructed. Loops often play crucial roles in protein function and can influence ligand binding by providing flexibility or forming important interactions. Modeling missing loops can be challenging, as they often exhibit higher flexibility and variability compared to other parts of the protein. Advanced loop modeling algorithms, such as those based on homology modeling or de novo structure prediction, can be utilized to generate plausible loop conformations.
However, it is important to exercise caution when dealing with regions of the protein structure that are weakly defined by experimental observables. These regions may have low occupancy, high displacement parameters (B values), or poor electron density in the crystallographic data. In such cases, the modeled side chains or loops should be examined critically, as their conformations may be less reliable. It may be necessary to consider alternative conformations or to treat these regions with a higher degree of flexibility during the docking process.
Water molecules
When preparing protein structures for docking studies, the treatment of water molecules is a critical consideration that can significantly impact the accuracy of the results. Water molecules can play various roles in protein-ligand binding, and a careful analysis is necessary to determine which water molecules should be included or excluded in the docking process.
In some cases, water molecules can mediate important interactions between the protein and ligand by forming bridging hydrogen bonds that stabilize the binding. Excluding such bridging water molecules from the docking procedure could lead to incorrect predictions of ligand poses and binding modes. For example, in docking studies of EGFR kinase inhibitors, researchers found that including a key bridging water molecule was essential for correctly reproducing the experimentally observed binding modes. When this water was excluded, the docking results yielded incorrect ligand poses.
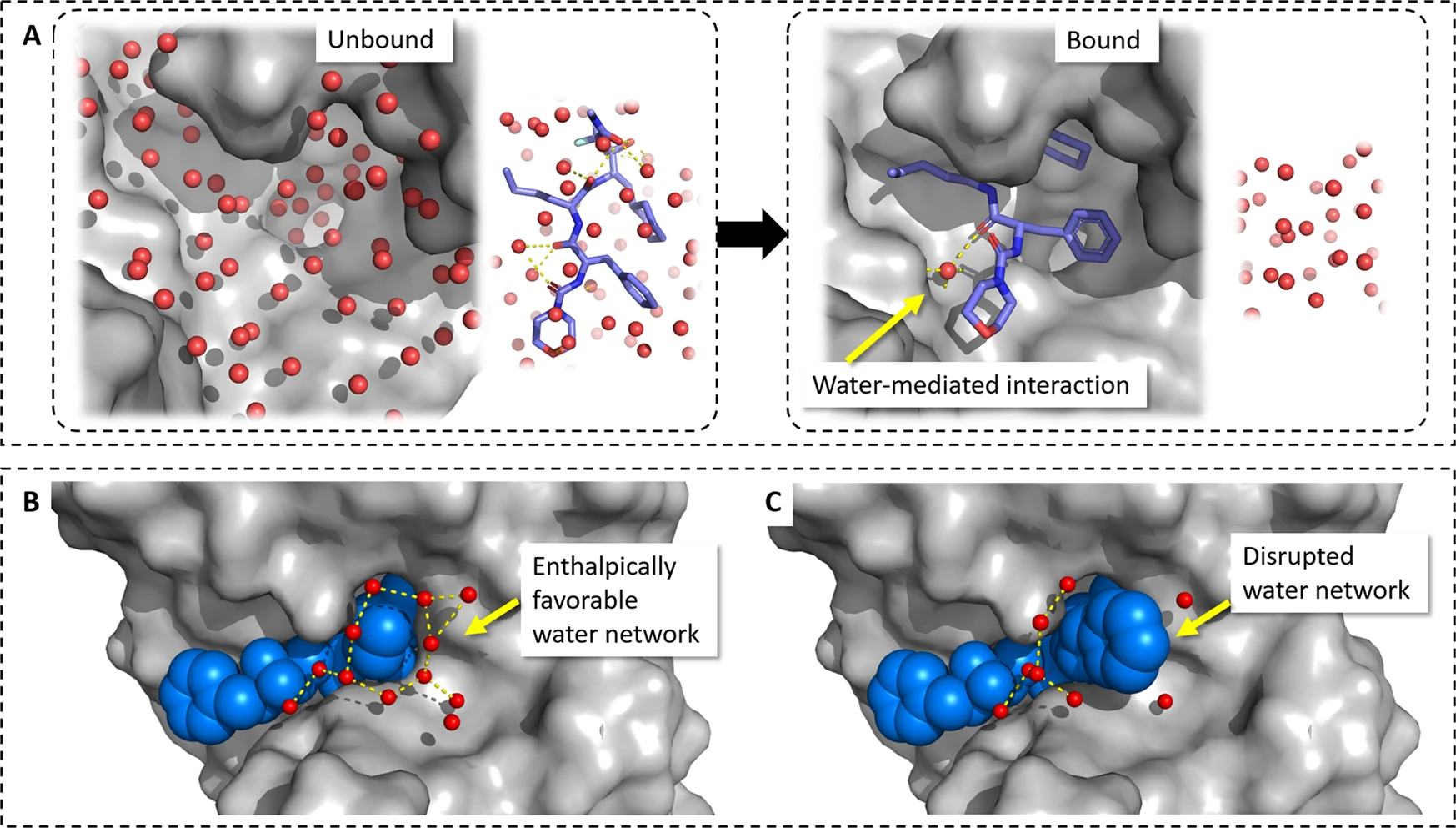
On the other hand, some water molecules may need to be displaced upon ligand binding. If a water molecule is energetically unfavorable or prevents optimal ligand contacts, it will likely be replaced by the ligand during the binding process. Including such water molecules in the docking procedure could hinder the identification of the correct ligand pose.
To make informed decisions about which water molecules to include or exclude, a careful examination of the available data is necessary. Crystallographic evidence of strongly bound water molecules, particularly those that form multiple hydrogen bonds with the protein and/or ligand, suggests that they should be retained in the docking process. Conversely, water molecules with high B-factors or incomplete electron density are less likely to be critical for ligand binding and can often be safely removed.
Computational methods can provide valuable insights into the thermodynamic properties of binding site water molecules. For example, the WATsite approach, which utilizes molecular dynamics simulations, can help identify the energetic favorability of individual water molecules in the binding site. This information can guide decisions on which water molecules should be included as part of the protein structure for docking.
Buffers
It is important to consider the presence of buffer components such as polyethylene glycol (PEG) and salts. These components are often used in protein crystallization to promote the formation of well-ordered crystals suitable for X-ray diffraction experiments. However, these molecules are typically not physiologically relevant to the protein's function and are specific to the crystallization conditions.
In most cases, buffer components like PEG and salts should be removed from the protein structure before conducting docking studies. These molecules may interfere with the docking process by occupying potential ligand binding sites or by altering the electrostatic properties of the protein surface. Removing these components allows for a clearer assessment of the ligand-protein interactions without the influence of artifacts introduced by the crystallization conditions.
There are some exceptions to this general rule. In rare cases, buffer components may mimic the interactions of a ligand or play a role in stabilizing a particular protein conformation relevant to ligand binding. In such instances, it may be appropriate to retain these molecules in the docking study. However, these cases are uncommon, and the decision to include buffer components should be based on a careful analysis of their potential impact on the ligand binding process.
Cofactors
The accuracy of docking results can be significantly influenced by the presence of cofactors, such as heme groups or metal ions, which are often essential for ligand recognition and binding. Cofactors are non-protein chemical compounds that associate with proteins and are necessary for their biological activity. In many cases, cofactors directly participate in ligand binding by forming interactions with the ligand or by inducing conformational changes in the protein that facilitate ligand binding. Therefore, the inclusion of cofactors in protein-ligand docking can be crucial for obtaining reliable and biologically relevant results.
When deciding whether to include cofactors in a docking study, several factors should be considered.
- The biological relevance of the cofactor must be assessed. If the cofactor is known to be involved in ligand recognition or is located in the binding site, it should be included in the docking process. Omitting biologically relevant cofactors may lead to incorrect binding poses and inaccurate affinity predictions.
- the availability of high-quality structural data for the protein-cofactor complex is essential. X-ray crystallography or NMR structures that include the cofactor provide the necessary information for accurate docking.
- the chosen docking software should support the inclusion of cofactors. Some docking programs have specific features for handling cofactors, such as the ability to define custom atom types or to specify the coordination geometry of metal ions. Finally, including cofactors in docking simulations can increase the computational complexity and resource requirements. The additional atoms and potential interactions introduced by the cofactor may lead to longer computation times and higher memory usage.
In cases where the cofactor is not directly involved in ligand binding or is located far from the binding site, its inclusion may not be necessary. However, if there is evidence that the cofactor induces long-range conformational changes in the protein that affect ligand binding, it should be considered in the docking process. In conclusion, the decision to include cofactors in protein-ligand docking should be based on a careful evaluation of their biological relevance, the availability of structural data, the capabilities of the docking software, and the computational resources at hand. When cofactors play a significant role in ligand recognition, their inclusion is essential for obtaining accurate and reliable docking results.
Protonation states
-
Bender, B. J., Gahbauer, S., Luttens, A., Lyu, J., Webb, C. M., Stein, R. M., ... & Shoichet, B. K. (2021). A practical guide to large-scale docking. Nature protocols, 16(10), 4799-4832. DOI: 10.1038/s41596-021-00597-z ↩
Ligand ↵
Ligand
TODO:
Conformation search
TODO:
Ended: Ligand
Ended: Preparation
Docking types ↵
Docking types
TODO:
Rigid docking
TODO:
Semiflexible docking
TODO:
Flexible docking
TODO:
Ended: Docking types
Scoring functions ↵
Scoring functions
TODO:
-
Zheng, L., Meng, J., Jiang, K., Lan, H., Wang, Z., Lin, M., ... & Mu, Y. (2022). Improving protein–ligand docking and screening accuracies by incorporating a scoring function correction term. Briefings in Bioinformatics, 23(3), bbac051. ↩
-
Shen, C., Ding, J., Wang, Z., Cao, D., Ding, X., & Hou, T. (2020). From machine learning to deep learning: Advances in scoring functions for protein–ligand docking. Wiley Interdisciplinary Reviews: Computational Molecular Science, 10(1), e1429. DOI: 10.1093/bib/bbac051 ↩
Physics ↵
Physics
TODO:
Autodock ↵
AutoDock2
AutoDock21 from 1996 is designed to predict the bound conformations of small, flexible ligands to macromolecular targets of known structure. This version combines simulated annealing for conformational searching with a rapid, grid-based method of energy evaluation using the AMBER force field.
Components
The scoring function in AutoDock2 estimates the binding free energy change (\(\Delta G_{binding}\)) using several energy terms that account for different types of interactions between the ligand and the protein. The total binding free energy change is the sum of all individual energy terms:
This empirical scoring function enables AutoDock2 to predict the binding affinity of a ligand to a protein by evaluating the sum of these interactions.
Van der Waals
The van der Waals interactions are modeled using a Lennard-Jones 12-6 potential, which captures both attractive (dispersion) and repulsive interactions between non-bonded atoms:
where \(A_{ij}\) and \(B_{ij}\) are parameters for atom pair \(i, j\), and \(r_{ij}\) is the distance between atoms \(i\) and \(j\). These parameters are essential for accurately modeling the attractive and repulsive forces between non-bonded atoms.
The parameters \(A_{ij}\) and \(B_{ij}\) are calculated using the following relationships, which are based on the well depth (\(\epsilon_{ij}\)) and the equilibrium contact distance (\(r_{eq,ij}\)) for each pair of atoms:
- Equilibrium Contact Distance (\(r_{eq}\)): The equilibrium contact distance \(r_{eq}\) is the distance at which the potential energy is at its minimum, indicating the optimal distance for van der Waals interactions.
- Well Depth (\(\epsilon\)): The well depth \(\epsilon\) represents the depth of the potential well, indicating the strength of the attractive interaction at the equilibrium distance.
Fitted parameters
| Atom Type | Van der Waals Radius (Å) | Van der Waals Well Depth (kcal/mol) |
|---|---|---|
| C | 2.00 | 0.15 |
| P | 2.10 | 0.20 |
| N | 1.75 | 0.16 |
| O | 1.60 | 0.20 |
| S | 2.00 | 0.20 |
| H | 1.00 | 0.02 |
The relationships for calculating \(A\) and \(B\) are:
These formulas ensure that the potential has a minimum value of \(-\epsilon_{ij}\) at the equilibrium distance \(r_{eq,ij}\).
Mixing rules
For heterogeneous pairs of atoms (i.e., atoms of different types), the \(\epsilon\) and \(r_{eq}\) values are calculated using combining rules:
-
Geometric Mean for Well Depth:
\[\epsilon_{ij} = \sqrt{\epsilon_{ii} \epsilon_{jj}}\] -
Arithmetic Mean for Equilibrium Distance:
\[r_{eq,ij} = \frac{r_{eq,ii} + r_{eq,jj}}{2}\]
These combining rules provide a way to estimate the interaction parameters for pairs of different atom types based on the parameters for homogeneous pairs.
Example
Suppose we have two atom types with the following parameters:
- Atom type 1: \(\epsilon_{11} = 0.15\) kcal/mol, \(r_{eq,11} = 2.00\) Å
- Atom type 2: \(\epsilon_{22} = 0.20\) kcal/mol, \(r_{eq,22} = 2.10\) Å
First, compute the combined parameters for the heterogeneous pair (1-2):
-
Well depth (\(\epsilon_{12}\)):
\[ \epsilon_{12} = \sqrt{0.15 \times 0.20} = \sqrt{0.03} \approx 0.173 \text{ kcal/mol} \] -
Equilibrium contact distance (\(r_{eq,12}\)):
\[ r_{eq,12} = \frac{2.00 + 2.10}{2} = 2.05 \text{ Å} \]
Then, calculate the \(A_{12}\) and \(B_{12}\) parameters:
-
\(A_{12}\):
\[ A_{12} = 4 \times 0.173 \times (2.05)^{12} \approx 4 \times 0.173 \times 458.485 \approx 317.43 \text{ kcal·Å}^{12}/\text{mol} \] -
\(B_{12}\):
\[ B_{12} = 4 \times 0.173 \times (2.05)^{6} \approx 4 \times 0.173 \times 114.89 \approx 79.51 \text{ kcal·Å}^{6}/\text{mol} \]
These values of \(A\) and \(B\) can now be used in the Lennard-Jones 12-6 potential to model the van der Waals interactions between atom type 1 and atom type 2.
Hydrogen Bonding
Hydrogen bonds are represented using a 12-10 potential, which emphasizes the directionality of hydrogen bonds:
Here, \(C_{ij}\) and \(D_{ij}\) are hydrogen bond parameters, and \(r_{ij}\) is the distance between the donor and acceptor atoms. These parameters are also derived from equilibrium distances and well depths.
Fitted parameters
| Atom Type | Hydrogen Bond Radius (Å) | Hydrogen Bond Well Depth (kcal/mol) |
|---|---|---|
| N | 1.9 | 5.0 |
| O | 1.9 | 5.0 |
| S | 2.5 | 1.0 |
The parameters \(C_{ij}\) and \(D_{ij}\) are calculated using the following relationships, which are based on the well depth (\(\epsilon_{ij}\)) and the equilibrium contact distance (\(r_{eq,ij}\)) for each pair of hydrogen-bonded atoms. The relationships for calculating \(C\) and \(D\) are:
These formulas ensure that the potential has a minimum value of \(-\epsilon_{ij}\) at the equilibrium distance \(r_{eq,ij}\).
Electrostatics
Electrostatic interactions are calculated using a Coulomb potential with a distance-dependent dielectric function:
where:
- \(q_i\) and \(q_j\) are the partial charges on atoms \(i\) and \(j\), respectively.
- \(r_{ij}\) is the distance between these atoms.
- \(\epsilon(r_{ij})\) is the dielectric constant, which varies as a function of the distance between the interacting charges.
Dielectric function
The electrostatic interactions between the ligand and the protein are a critical component of the scoring function. To accurately simulate these interactions in a solvated environment, AutoDock employs a sigmoidal distance-dependent dielectric function. This approach allows for a more realistic representation of solvent effects, which vary with distance, compared to a constant dielectric model.
In a biological context, the dielectric constant is not uniform; it changes with the distance due to the presence of water and other solvents. To account for this, AutoDock 2.4 uses a sigmoidal distance-dependent dielectric function, which provides a more accurate simulation of solvent effects. The function is defined as follows:
where:
- \(A\) is the dielectric constant close to the protein surface. This value typically represents the lower dielectric environment near the protein surface, reflecting the lower dielectric constant of the protein interior.
- \(B\) is the dielectric constant in the bulk solvent (e.g., water). This is the dielectric constant of the solvent, usually around 78.4 for water at 25°C.
- \(k\) is a parameter that determines the steepness of the sigmoid curve. This parameter controls how quickly the dielectric constant transitions from \(A\) to \(B\) as the distance increases.
- \(r\) is the distance between the interacting atoms.
- \(r_0\) is the midpoint distance where the dielectric constant is halfway between \(A\) and \(B\). This distance defines the point at which the dielectric constant is the average of \(A\) and \(B\).
The sigmoidal function offers several advantages over a constant dielectric model:
- Realistic Solvent Effects: It provides a smooth transition from the low dielectric environment near the protein surface to the high dielectric environment of the bulk solvent, accurately reflecting the physical environment.
- Improved Accuracy: By varying the dielectric constant with distance, it captures the screening effect of the solvent more precisely, leading to better predictions of electrostatic interactions and binding affinities.
- Flexibility: The parameters \(A\), \(B\), \(k\), and \(r_0\) can be adjusted to model different solvent environments and conditions, enhancing the versatility of the docking simulations.
Internal Ligand Energy
The internal energy of the ligand includes contributions from van der Waals interactions, hydrogen bonds, and electrostatic interactions within the ligand itself. This term ensures that the ligand maintains a realistic conformation during docking:
Evaluation
AutoDock2 employs a grid-based method to precalculate and store interaction energies for rapid evaluation during docking. This method involves creating three-dimensional grid maps of pairwise atomic interaction energies around the protein target, allowing for efficient and accurate energy calculations during the docking simulation.
Precalculation of Grid Maps
The process of generating interaction maps in AutoDock2 involves several key steps:
- Protein Target Placement: The protein target is placed in a three-dimensional grid. Each grid point represents a position in space around the protein.
- Probe Atom Evaluation: A probe atom systematically visits each grid point. For each grid point, the pairwise interaction energy of the probe atom with all protein atoms within a cutoff radius of 8 Å is calculated and summed. These interactions include van der Waals forces, hydrogen bonding, and electrostatic interactions.
- Storage of Interaction Energies: The calculated interaction energies for each grid point are stored in separate grid maps, one for each type of atom in the ligand. This results in multiple grid maps, each corresponding to different interaction types such as dispersion/repulsion, hydrogen bonding, and electrostatic potential.
Grid Sampling
Once the grid maps are precalculated, they are loaded into memory at the beginning of the docking process. These grid maps contain precomputed interaction energies at discrete points in a three-dimensional space surrounding the protein target. During the docking simulation, as the ligand explores different conformations and positions, the interaction energy of each ligand atom with the protein is determined by referencing these grid maps.
To evaluate the interaction energy at any given point in space, the ligand's atoms sample the surrounding grid points. This is achieved through trilinear interpolation, a method that interpolates values within a three-dimensional grid. Trilinear interpolation involves considering the eight grid points that form a cube around the point of interest. The energy values at these eight points are combined based on their distances to the actual position of the ligand atom, providing a smooth and continuous estimate of the interaction energy.
This interpolation process allows for rapid and efficient calculation of the ligand's binding energy, as it avoids the need to compute the interaction energy from scratch at every step. Instead, it leverages the precomputed grid values, significantly speeding up the docking simulation while maintaining accuracy. By using trilinear interpolation, AutoDock2 can swiftly evaluate numerous conformations and positions of the ligand, facilitating an efficient exploration of the binding landscape.
Ligand intramolecular contributions
In addition to evaluating the intermolecular interactions between the ligand and the protein, AutoDock2 also considers the intramolecular interactions within the ligand itself. This is crucial for maintaining realistic ligand conformations during the docking process. The intramolecular evaluation ensures that the ligand does not adopt physically improbable conformations that would lead to inaccurate binding predictions.
Intramolecular energies are calculated at each step of the docking simulation to ensure that the ligand maintains a realistic conformation. The steps involved in calculating intramolecular energies are:
- Torsion Angle Adjustments: Changes in the ligand's torsional angles are applied, allowing the ligand to explore different conformations.
- Energy Calculations: The intramolecular van der Waals, hydrogen bonding, and electrostatic energies are calculated based on the current conformation of the ligand.
- Conformation Evaluation: The total intramolecular energy is evaluated to determine if the current conformation is physically plausible. High intramolecular energy indicates unfavorable conformations, which are less likely to be accepted during the simulated annealing process.
By incorporating intramolecular evaluation, AutoDock2 ensures that the ligand adopts conformations that are energetically favorable and realistic. This leads to more accurate predictions of binding modes and affinities, as the ligand's internal strain is minimized. The consideration of intramolecular energies also helps in distinguishing between different binding poses, favoring those that are not only favorable in terms of ligand-protein interactions but also in terms of internal ligand stability.
Calibration and Validation
The empirical coefficients for each term in the scoring function were calibrated using a set of known protein-ligand complexes. Linear regression analysis was performed to determine the coefficients, ensuring that the scoring function accurately reflects observed binding affinities. Validation was achieved by comparing predicted binding energies with experimental data, showing good correlation.
Fitting Details
The fitting of the scoring function involves determining the optimal parameters for each energy term to match experimental binding free energies. This process includes:
- Data Collection: Gathering a dataset of protein-ligand complexes with known binding constants.
- Parameter Estimation: Using regression techniques to estimate parameters such as \(A_{ij}\), \(B_{ij}\), \(C_{ij}\), \(D_{ij}\), and partial charges.
- Optimization: Iteratively adjusting parameters to minimize the difference between predicted and experimental binding free energies.
- Validation: Comparing the fitted scoring function against a separate validation set to ensure its accuracy and generalizability.
-
Morris, G. M., Goodsell, D. S., Huey, R., & Olson, A. J. (1996). Distributed automated docking of flexible ligands to proteins: parallel applications of AutoDock2. Journal of computer-aided molecular design, 10, 293-304. DOI: 10.1007/BF00124499 ↩
AutoDock3
AutoDock31 from 1998 is a powerful tool used in computational drug design for predicting the binding conformations of flexible ligands to macromolecular targets. The success of AutoDock3 is largely due to its scoring function, which estimates the free energy change upon binding of the ligand to the protein. This section will provide a detailed explanation of the scoring function used in AutoDock3, focusing on its components and their contributions to the overall binding free energy.
Components
The scoring function in AutoDock3 is an empirical free energy function that has been calibrated using a set of 30 protein-ligand complexes with known binding constants. The function is designed to predict the binding free energy change (\(\Delta G_{binding}\)) and is composed of several terms that account for different types of interactions between the ligand and the protein.
The total free energy change upon binding is the sum of all these individual terms:
This empirical scoring function allows AutoDock3 to predict the binding affinity of a ligand to a protein by evaluating the sum of these contributions, thus providing insights into the strength and nature of the interaction.
Van der Waals
The van der Waals interactions are described using a Lennard-Jones 12-6 potential, which accounts for both attractive (dispersion) and repulsive interactions between non-bonded atoms. The equation for the van der Waals term is:
where \(A_{ij}\) and \(B_{ij}\) are the van der Waals parameters for atom pair \(i, j\), and \(r_{ij}\) is the distance between atoms \(i\) and \(j\).
Hydrogen Bonding
Hydrogen bonds are modeled using a directional 12-10 potential, which emphasizes the angle dependency of hydrogen bonds, ensuring that only properly aligned hydrogen bonds contribute significantly to the energy:
Here, \(C_{ij}\) and \(D_{ij}\) are parameters specific to the hydrogen bond interaction, and \(E(\theta)\) is an angular term that ensures directionality.
Electrostatic
Electrostatic interactions are calculated using a Coulomb potential with a distance-dependent dielectric function to account for the screening effect in a solvated environment:
where \(q_i\) and \(q_j\) are the partial charges on atoms \(i\) and \(j\), respectively, and \(\epsilon(r_{ij})\) is the dielectric constant as a function of distance.
Torsional
The torsional free energy term accounts for the loss of rotational freedom of the ligand upon binding and is proportional to the number of rotatable bonds in the ligand:
where \(N_{tor}\) is the number of rotatable bonds, and \(k_{tor}\) is an empirically determined constant.
Desolvation
The desolvation term estimates the free energy change associated with the removal of the ligand from the solvent and its burial in the binding pocket of the protein. It includes contributions from both polar and nonpolar atoms:
where \(S_{i,j}\) are solvation parameters, \(V_{j}\) is the volume of the protein atom \(j\), and \(\sigma\) is a parameter related to the solvation shell thickness.
Calibration and Validation
The empirical coefficients for each term in the scoring function were determined through linear regression analysis using a dataset of known protein-ligand complexes.
Protein-ligand complex table
| Protein–Ligand Complex | PDB Code | Log Ki |
|---|---|---|
| Concanavalin A / α-methyl-D-mannopyranoside | 4cna | 2 |
| Carboxypeptidase A / glycyl-L-tyrosine | 3cpa | 3.88 |
| Carboxypeptidase A / phosphonate ZAA(P=O)P(O)2 | 6cpa | 11.52 |
| Cytochrome P-450 / camphor | 2cpp | 6.07 |
| Dihydrofolate reductase / methotrexate | 4dfr | 9.7 |
| α-Thrombin / benzamidine | 1dwb | 2.92 |
| Endothiapepsin / H-256 | zer6 | 7.22 |
| ε-Thrombin / MQPA | 1etr | 7.4 |
| ε-Thrombin / NAPAP | 1ets | 8.52 |
| ε-Thrombin / 4-TAPAP | 1ett | 6.19 |
| FK506-binding protein(FKBP) / immunosuppressant FK506 | 1fkf | 9.7 |
| D-Galactose / D-glucose binding protein / galactose | 2gbp | 7.6 |
| Hemagglutinin / sialic acid | 4hmg | 2.55 |
| HIV-1 Protease / A78791 | 1hvj | 10.46 |
| HIV-1 Protease / MVT101 | 4hvp | 6.15 |
| HIV-1 Protease / acylpepstatine | 5hvp | 5.96 |
| HIV-1 Protease / XK263 | 1hvr | 9.51 |
| Fatty-acid-binding protein / C15COOH | 2ifb | 5.43 |
| Myoglobin(ferric) / imidazole | 1mbi | 1.88 |
| McPC603 / phosphocholine | 2mcp | 5.23 |
| β-Trypsin / benzamidine | 3ptb | 4.74 |
| Retinol-binding protein / retinol | 1rbp | 6.72 |
| Thermolysin / Leu-hydroxylamine | 4tln | 3.72 |
| Thermolysin / phosphoramidon | 1tlp | 7.55 |
| Thermolysin / n-(1-carboxy-3-phenylpropyl)-Leu-Trp | 1tmn | 7.3 |
| Thermolysin / Cbz-Phe-p-Leu-Ala(ZFpLA) | 4tmn | 10.19 |
| Thermolysin / Cbz-Gly-p-Leu-Leu(ZGpLL) | 5tmn | 8.04 |
| Purine nucleoside phosphorylase(PNP) / guanine | 1ulb | 5.3 |
| Xylose isomerase / CB3717 | 2xis | 5.82 |
| Triose phosphate isomerase(TIM) / 2-phosphoglycolic acid(PGA) | 2ypi | 4.82 |
This calibration process ensures that the scoring function accurately reflects the observed binding affinities. The final model was validated by comparing its predictions with experimental binding constants, demonstrating good correlation and reliability.
Changes from AutoDock2
Similarities
The scoring functions of AutoDock2 and AutoDock3 share several core features. Both versions use a Lennard-Jones 12-6 potential to model van der Waals interactions. This potential captures both the attractive and repulsive forces between non-bonded atoms, and the parameters \(A_{ij}\) and \(B_{ij}\) are derived from well depths (\(\epsilon\)) and equilibrium contact distances (\(r_{eq}\)). Additionally, both versions employ a 12-10 potential to represent hydrogen bonding interactions. The parameters \(C_{ij}\) and \(D_{ij}\) for hydrogen bonds are similarly derived from the well depths and equilibrium distances, ensuring accurate modeling of the directionality and strength of hydrogen bonds.
Electrostatic interactions in both AutoDock2 and AutoDock3 are calculated using Coulomb's law, which describes the force between two charges. Both versions can use a distance-dependent dielectric function to simulate solvent effects, making the electrostatic interaction calculations more realistic by accounting for the varying dielectric environment in the presence of water and other solvents.
Changes
While the core components of the scoring function remain the same, AutoDock3 introduces several significant enhancements and new terms that improve the accuracy and reliability of docking predictions.
One of the key improvements in AutoDock3 is the enhanced electrostatic interaction model. AutoDock3 uses an improved sigmoidal distance-dependent dielectric function to simulate the transition from the low dielectric constant near the protein surface to the high dielectric constant in the bulk solvent. This enhancement provides a more realistic representation of solvent effects, improving the accuracy of electrostatic interaction calculations.
AutoDock3 also introduces a new desolvation term. This term accounts for the energetic cost of removing the ligand from the solvent and placing it into the binding pocket of the protein. The desolvation term is particularly important for accurately predicting binding affinities, especially for hydrophobic interactions where the removal of water molecules plays a crucial role.
Another significant addition in AutoDock3 is the torsional free energy term. This term accounts for the loss of rotational entropy of the ligand upon binding to the protein. The torsional free energy term is proportional to the number of rotatable bonds in the ligand, penalizing flexible ligands that lose more entropy upon binding. This addition improves the prediction of binding affinities for ligands with many rotatable bonds, making the docking results more reliable.
-
Morris, G. M., Goodsell, D. S., Halliday, R. S., Huey, R., Hart, W. E., Belew, R. K., & Olson, A. J. (1998). Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. Journal of computational chemistry, 19(14), 1639-1662. DOI: 10.1002/(SICI)1096-987X(19981115)19:14<1639::AID-JCC10>3.0.CO;2-B ↩
AutoDock4
TODO:
-
Huey, R., Morris, G. M., Olson, A. J., & Goodsell, D. S. (2007). A semiempirical free energy force field with charge‐based desolvation. Journal of computational chemistry, 28(6), 1145-1152. DOI: 10.1002/jcc.20634 ↩
Ended: Autodock
Ended: Physics
Empirical
TODO:
.y Knowledge
TODO:
.y Machine learning
TODO:
Ended: Scoring functions
Pose refinement ↵
Pose refinement
TODO:
Molecular-dynamics based
TODO:
-
Guterres, H., & Im, W. (2020). Improving protein-ligand docking results with high-throughput molecular dynamics simulations. Journal of Chemical Information and Modeling, 60(4), 2189-2198. DOI: 10.1021/acs.jcim.0c00057 ↩
Ended: Pose refinement
Packages ↵
Packages
TODO:
AutoDock4
TODO:
-
Morris, G. M., Huey, R., Lindstrom, W., Sanner, M. F., Belew, R. K., Goodsell, D. S., & Olson, A. J. (2009). AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of computational chemistry, 30(16), 2785-2791. DOI: 10.1002/jcc.21256 ↩
-
Santos-Martins, D., Solis-Vasquez, L., Tillack, A. F., Sanner, M. F., Koch, A., & Forli, S. (2021). Accelerating AutoDock4 with GPUs and gradient-based local search. Journal of chemical theory and computation, 17(2), 1060-1073. DOI: 10.1021/acs.jctc.0c01006 ↩
-
Goodsell, D. S., Sanner, M. F., Olson, A. J., & Forli, S. (2021). The AutoDock suite at 30. Protein Science, 30(1), 31-43. DOI: 10.1002/pro.3934 ↩
DOCK 6
TODO:
-
Allen, W. J., Balius, T. E., Mukherjee, S., Brozell, S. R., Moustakas, D. T., Lang, P. T., ... & Rizzo, R. C. (2015). DOCK 6: Impact of new features and current docking performance. Journal of computational chemistry, 36(15), 1132-1156. DOI: 10.1002/jcc.23905 ↩
Glide
TODO:
-
Friesner, R. A., Banks, J. L., Murphy, R. B., Halgren, T. A., Klicic, J. J., Mainz, D. T., ... & Shenkin, P. S. (2004). Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. Journal of medicinal chemistry, 47(7), 1739-1749. DOI: 10.1021/jm0306430 ↩
GOLD
TODO:
-
Verdonk, M. L., Cole, J. C., Hartshorn, M. J., Murray, C. W., & Taylor, R. D. (2003). Improved protein–ligand docking using GOLD. Proteins: Structure, Function, and Bioinformatics, 52(4), 609-623. DOI: 10.1002/prot.10465 ↩
AutoDock Vina
TODO:
-
Eberhardt, J., Santos-Martins, D., Tillack, A. F., & Forli, S. (2021). AutoDock Vina 1.2.0: New docking methods, expanded force field, and python bindings. Journal of chemical information and modeling, 61(8), 3891-3898. DOI: 10.1021/acs.jcim.1c00203 ↩
-
Trott, O., & Olson, A. J. (2010). AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of computational chemistry, 31(2), 455-461. DOI: 10.1002/jcc.21334 ↩
Glycans ↵
Glycan packages
TODO:
GlycanDock
TODO:
-
Nance, M. L., Labonte, J. W., Adolf-Bryfogle, J., & Gray, J. J. (2021). Development and Evaluation of GlycanDock: a protein–glycoligand docking refinement algorithm in Rosetta. Journal of Physical Chemistry B, 125(25), 6807-6820. DOI: 10.1021/acs.jpcb.1c00910 ↩
GlycoTorch vina
TODO:
-
Boittier, E. D., Burns, J. M., Gandhi, N. S., & Ferro, V. (2020). GlycoTorch Vina: docking designed and tested for glycosaminoglycans. Journal of Chemical Information and Modeling, 60(12), 6328-6343. DOI: 10.1021/acs.jcim.0c00373 ↩
Vina-Carb
TODO:
-
Nivedha, A. K., Thieker, D. F., Makeneni, S., Hu, H., & Woods, R. J. (2016). Vina-Carb: improving glycosidic angles during carbohydrate docking. Journal of chemical theory and computation, 12(2), 892-901. DOI: 10.1021/acs.jctc.5b00834 ↩
Ended: Glycans
Ended: Packages
Ended: Docking
Molecular simulations ↵
Molecular simulations
TODO:
Molecular mechanics ↵
Molecular mechanics
Molecular mechanics is a computational approach that uses classical physics to model the potential energy of a molecular system. It provides a mathematical framework for understanding the energetics of molecules and their interactions. In molecular mechanics, the potential energy of a system is calculated using a force field, which is a set of equations and parameters that describe the various components contributing to the overall energy. These components include bond stretching, angle bending, torsional terms, and non-bonded interactions. By understanding the principles of molecular mechanics, we can gain valuable insights into the behavior of molecules and their potential as drug candidates.
TODO:
-
Chapter 6 of Kumar, T. D. A. (2022). Drug Design: A Conceptual Overview. CRC Press. DOI: 10.1201/9781003298755 ↩
-
Braun, E., Gilmer, J., Mayes, H. B., Mobley, D. L., Monroe, J. I., Prasad, S., & Zuckerman, D. M. (2018). Best Practices for Foundations in Molecular Simulations [Article v1.0]. Living Journal of Computational Molecular Science, 1(1), 5957. DOI: 10.33011/livecoms.1.1.5957 ↩
-
Chapter 2 of Jensen, F. (2017). Introduction to computational chemistry. John wiley & sons. ↩
-
Chapter 2 of Cramer, C. J. (2013). Chapter 2 of Essentials of computational chemistry: Theories and models. John Wiley & Sons. ↩
-
Chapter 4 of Leach, A. R. (2001). Molecular modelling: Principles and applications. Pearson education. ↩
Force fields
TODO:
Bonded ↵
Covalent interactions
Covalent interactions are the chemical bonds formed by the sharing of electrons between atoms. These strong interactions play a crucial role in determining the structure, stability, and reactivity of molecules. In the context of drug design, covalent interactions are essential for understanding the binding of drugs to their targets. Covalent drugs, which form irreversible bonds with their targets, have unique advantages and disadvantages compared to non-covalent drugs. By exploring the nature of covalent interactions and their role in drug-target binding, we can design more effective and specific therapeutic agents.
TODO:
Bonds
TODO:
Angles
TODO:
Dihedrals
TODO:
Ended: Bonded
Nonbonded ↵
Nonbonded
TODO:
Electrostatic
TODO:
One common method for assigning partial charges is the Restrained ElectroStatic Potential (RESP) fitting scheme, which adds a penalty term to prevent unrealistically large charges on buried atoms. This approach has been used in popular force fields such as AMBER. Other constraints, such as equalizing charges on equivalent atoms or ensuring charge neutrality within subgroups, are also often imposed during the fitting process.
It is important to note that partial charges obtained from ESP fitting do not include the effects of polarization, which is the redistribution of electron density in response to an external electric field. Polarization is a many-body effect that arises from the interaction between two or more molecules and can significantly contribute to the overall electrostatic energy. Neglecting polarization can lead to a systematic underestimation of the electrostatic energy.
To account for polarization effects, polarizable force fields have been developed, which include additional terms to describe the induced dipole moments of atoms in response to the electric field generated by the surrounding atoms. These force fields provide a more accurate description of electrostatic interactions, particularly in systems with significant polarization effects, such as proteins and polar solvents.
The accuracy of electrostatic interactions in molecular simulations depends on several factors, including the quality of the partial charges, the treatment of polarization effects, and the choice of dielectric constant. In practice, the dielectric constant is often used as a scaling factor to implicitly account for the screening of electrostatic interactions by the surrounding environment, such as solvent molecules.
Van der Waals
TODO:
Ended: Nonbonded
Parameterization
TODO:
Ended: Molecular mechanics
Ended: Molecular simulations
Ended: Structure based
Ligand based ↵
Ligand-based drug design
TODO:
Context
Why are we here?
TODO:
Where are we now?
TODO:
Learning objectives
TODO:
Descriptors ↵
Descriptors
TODO:
-
Chapter 5 of Kumar, T. D. A. (2022). Drug Design: A Conceptual Overview. CRC Press. DOI: 10.1201/9781003298755 ↩
.y Pharmacophore
TODO:
-
Chapter 6 of Rudrapal, M., & Egbuna, C. (Eds.). (2022). Computer aided drug design (CADD): From ligand-based methods to structure-based approaches. Elsevier. ↩
Ended: Descriptors
QSAR
TODO:
-
Chapter 4 of Rudrapal, M., & Egbuna, C. (Eds.). (2022). Computer aided drug design (CADD): From ligand-based methods to structure-based approaches. Elsevier. ↩
-
Chapter 5 of Kumar, T. D. A. (2022). Drug Design: A Conceptual Overview. CRC Press. DOI: 10.1201/9781003298755 ↩
Virtual screening
TODO:
ADMET
TODO:
Ended: Ligand based
Advanced topics ↵
Advanced topics in CADD
TODO:
Context
Why are we here?
TODO:
Where are we now?
TODO:
Learning objectives
TODO:
Lead optimization
TODO:
Ended: Advanced topics
Case studies
TODO:
Context
Why are we here?
TODO:
Where are we now?
TODO:
Learning objectives
TODO:
Appendices ↵
Appendices
TODO:
References ↵
References
The following resources are recommended for further reading.
Textbooks
- Rosa, J. M. C. (2023). Pharmaceutical chemistry: Drug design and action. Walter de Gruyter GmbH & Co KG.
- 📚 Kumar, T. D. A. (2022). Drug design: A conceptual overview. CRC Press. DOI: 10.1201/9781003298755
- Rudrapal, M., & Egbuna, C. (Eds.). (2022). Computer aided drug design (CADD): From ligand-based methods to structure-based approaches. Elsevier.
- 📚 Renaud, J.-P. (Eds.). (2020). Structural biology in drug discovery: Methods, techniques, and practices. John Wiley & Sons.
- Strømgaard, K., Krogsgaard-Larsen, P., Madsen, U. (2017). Textbook of drug design and discovery. CRC Press.
- 📚 Jensen, F. (2017). Introduction to computational chemistry. John Wiley & Sons.
- Cooksy, A. (2014). Physical chemistry: Thermodynamics, statistical mechanics, and kinetics. Pearson.
- Cooksy, A. (2014). Physical chemistry: Quantum chemistry and molecular interactions. Pearson.
- 📚 Stone, A. J. (2013). The theory of intermolecular forces. Oxford University Press.
- Cramer, C. J. (2013). Chapter 2 of Essentials of computational chemistry: Theories and models. John Wiley & Sons.
- Phillips, R., Kondev, J., Theriot, J., & Garcia, H. (2012). Physical biology of the cell. Garland Science.
- 📚 Zuckerman, D. M. (2010). Statistical physics of biomolecules: An introduction. CRC Press.
- Anslyn, E. V., & Dougherty, D. A. (2006). Modern physical organic chemistry. University Science Books.
- Leach, A. R. (2001). Molecular modelling: Principles and applications. Pearson Education.
Note
📚 means this textbook is freely available online through the University of Pittsburgh library.
Literature ↵
Literature
TODO:
AutoDock Vina Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading (@trott2009autodock)
Title: AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading
Authors: Oleg Trott, Arthur J. Olson
Journal: Journal of Computational Chemistry
Year: 2009
DOI: 10.1002/jcc.21334
Abstract
AutoDock Vina, a new program for molecular docking and virtual screening, is presented. AutoDock Vina achieves an approximately two orders of magnitude speed-up compared with the molecular docking software previously developed in our lab (AutoDock 4), while also significantly improving the accuracy of the binding mode predictions, judging by our tests on the training set used in AutoDock 4 development. Further speed-up is achieved from parallelism, by using multithreading on multicore machines. AutoDock Vina automatically calculates the grid maps and clusters the results in a way transparent to the user.
Takeaways
- TODO: Add takeaways if desired.
Main
TODO: Add notes if desired
Bibtex
@article{trott2010autodock,
title={AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading},
author={Trott, Oleg and Olson, Arthur J},
journal={Journal of Computational Chemistry},
volume={31},
number={2},
pages={455--461},
year={2010},
publisher={Wiley Online Library}
}
AutoDock vina 1.2.0 New docking methods, expanded force field, and python bindings (@eberhardt2021autodock)
Title: AutoDock vina 1.2.0: New docking methods, expanded force field, and python bindings
Authors: Jerome Eberhardt, Diogo Santos-Martins, Andreas F. Tillack, Stefano Forli
Journal: Journal of Chemical Information and Modeling
Year: 2021
Abstract
AutoDock Vina is arguably one of the fastest and most widely used open-source programs for molecular docking. However, compared to other programs in the AutoDock Suite, it lacks support for modeling specific features such as macrocycles or explicit water molecules. Here, we describe the implementation of this functionality in AutoDock Vina 1.2.0. Additionally, AutoDock Vina 1.2.0 supports the AutoDock4.2 scoring function, simultaneous docking of multiple ligands, and a batch mode for docking a large number of ligands. Furthermore, we implemented Python bindings to facilitate scripting and the development of docking workflows. This work is an effort toward the unification of the features of the AutoDock4 and AutoDock Vina programs.
Takeaways
- TODO: Add takeaways if desired.
Main
TODO: Add notes if desired
Bibtex
@article{eberhardt2021autodock,
title={AutoDock Vina 1.2.0: New docking methods, expanded force field, and python bindings},
author={Eberhardt, Jerome and Santos-Martins, Diogo and Tillack, Andreas F and Forli, Stefano},
journal={Journal of Chemical Information and Modeling},
volume={61},
number={8},
pages={3891--3898},
year={2021},
publisher={ACS Publications}
}
Automated docking of substrates to proteins by simulated annealing (@goodsell1990automated)
Title: Automated docking of substrates to proteins by simulated annealing
Authors: David S. Goodsell, Arthur J. Olson
Journal: Proteins: Structure, Function, and Bioinformatics
Year: 1990
Abstract
The Metropolis technique of conformation searching is combined with rapid energy evaluation using molecular affinity potentials to give an efficient procedure for docking substrates to macromolecules of known structure. The procedure works well on a number of crystallographic test systems, functionally reproducing the observed binding modes of several substrates.
Takeaways
- TODO: Add takeaways if desired.
Main
TODO: Add notes if desired
Bibtex
@article{goodsell1990automated,
title={Automated docking of substrates to proteins by simulated annealing},
author={Goodsell, David S and Olson, Arthur J},
journal={Proteins: Structure, Function, and Bioinformatics},
volume={8},
number={3},
pages={195--202},
year={1990},
publisher={Wiley Online Library}
}
Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function (@morris1998automated)
Title: Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function
Authors: Garrett M. Morris, David S. Goodsell, Robert S. Halliday, Ruth Huey, William E. Hart, Richard K. Belew, Arthur J. Olson
Journal: Journal of Computational Chemistry
Year: 1998
DOI: 10.1002/(sici)1096-987x(19981115)19:14<1639::aid-jcc10>3.0.co;2-b
Abstract
A novel and robust automated docking method that predicts the bound conformations of flexible ligands to macromolecular targets has been developed and tested, in combination with a new scoring function that estimates the free energy change upon binding. Interestingly, this method applies a Lamarckian model of genetics, in which environmental adaptations of an individual's phenotype are reverse transcribed into its genotype and become heritable traits (sic). We consider three search methods, Monte Carlo simulated annealing, a traditional genetic algorithm, and the Lamarckian genetic algorithm, and compare their performance in dockings of seven protein–ligand test systems having known three-dimensional structure. We show that both the traditional and Lamarckian genetic algorithms can handle ligands with more degrees of freedom than the simulated annealing method used in earlier versions of AUTODOCK, and that the Lamarckian genetic algorithm is the most efficient, reliable, and successful of the three. The empirical free energy function was calibrated using a set of 30 structurally known protein–ligand complexes with experimentally determined binding constants. Linear regression analysis of the observed binding constants in terms of a wide variety of structure-derived molecular properties was performed. The final model had a residual standard error of 9.11 kJ mol−1 (2.177 kcal mol−1) and was chosen as the new energy function. The new search methods and empirical free energy function are available in AUTODOCK, version 3.0.
Takeaways
- TODO: Add takeaways if desired.
Main
TODO: Add notes if desired
Bibtex
@article{morris1998automated,
title={Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function},
author={Morris, Garrett M and Goodsell, David S and Halliday, Robert S and Huey, Ruth and Hart, William E and Belew, Richard K and Olson, Arthur J},
journal={Journal of computational chemistry},
volume={19},
number={14},
pages={1639--1662},
year={1998},
publisher={Wiley Online Library}
}
Ended: Literature
Ended: References
Data ↵
Data
Binding MOAD
DEKOIS
BioLiP2
ChEMBL
Database of bioactive molecules containing information manually extracted from articles, approved drugs and clinical development reports.
-
Zdrazil, B., Felix, E., Hunter, F., Manners, E. J., Blackshaw, J., Corbett, S., ... & Leach, A. R. (2024). The ChEMBL Database in 2023: a drug discovery platform spanning multiple bioactivity data types and time periods. Nucleic acids research, 52(D1), D1180-D1192. DOI: 10.1093/nar/gkad1004 ↩
BindingDB
DUD-AD
DUD-E
LIT-PCBA
TODO:
-
Tran-Nguyen, V. K., Jacquemard, C., & Rognan, D. (2020). LIT-PCBA: an unbiased data set for machine learning and virtual screening. Journal of chemical information and modeling, 60(9), 4263-4273. DOI: 10.1021/acs.jcim.0c00155 ↩
PubChem
Database containing verified active and inactive compounds.
-
Kim, S., Chen, J., Cheng, T., Gindulyte, A., He, J., He, S., Li, Q., Shoemaker, B. A., Thiessen, P. A., Yu, B., Zaslavsky, L., Zhang, J., & Bolton, E. E. (2023). PubChem 2023 update. Nucleic Acids Res., 51(D1), D1373–D1380. DOI: 10.1093/nar/gkac956 ↩
PDBbind+
Ended: Data
Universal gas constant
The universal gas constant (\(R\)) emerges as a fundamental constant that relates the microscopic properties of a system (e.g., the motion and interactions of individual particles) to its macroscopic thermodynamic properties (e.g., temperature, pressure, and energy).
The universal gas constant is defined as the product of the Boltzmann constant (\(k_B\)) and the Avogadro constant (\(N_A\)):
where:
- \(k_B\) is the Boltzmann constant 1, which relates the average kinetic energy of particles to the absolute temperature. Its value is \(1.380649 \times 10^{-23}\) J/K.
- \(N_A\) is the Avogadro constant 2, which represents the number of particles (atoms or molecules) in one mole of a substance. Its value is \(6.02214076 \times 10^{23}\) mol-1.
Thus, the value of \(R\) is 8.31446261815324 J K -1 mol-1.
The Boltzmann constant (\(k_B\)) is a fundamental constant in statistical mechanics, as it provides a link between the microscopic energy states of a system and its temperature. It appears in the Boltzmann distribution, which describes the probability distribution of energy states in a system at thermal equilibrium:
where:
- \(P(E)\) is the probability of a system being in a state with energy \(E\);
- \(Z\) is the partition function, which is a sum over all possible energy states;
- \(T\) is the absolute temperature in Kelvin.
The universal gas constant (\(R\)) inherits its fundamental role in statistical mechanics from the Boltzmann constant. It appears in the ideal gas law, which can be derived using statistical mechanics principles:
where:
- \(P\) is the pressure of the gas;
- \(V\) is the volume of the gas;
- \(n\) is the number of moles of the gas;
- \(T\) is the absolute temperature.
In the context of the free energy of binding, the universal gas constant plays a crucial role in connecting the microscopic details of ligand-protein interactions to the macroscopic thermodynamic description. The equilibrium constant (\(K_{eq}\)) of a binding reaction can be related to the partition functions of the free and bound states of the ligand and protein, which in turn depend on the microscopic energy states accessible to the system.
The relationship between the Gibbs free energy of binding (\(\Delta G\)) and the equilibrium constant (\(K_{eq}\)) arises from the statistical mechanics treatment of the system, where the universal gas constant (\(R\)) acts as a scaling factor that relates the microscopic energy states to the macroscopic thermodynamic quantities.
Quantum chemistry data ↵
Quantum chemistry data
TODO:
Geometry optimization¶
from pyscf import gto, scf, mp
from pyscf.geomopt.geometric_solver import optimize
import py3Dmol
mol = gto.Mole()
mol.atom = """
H 1.090669 0.975611 0.000000
H -0.435544 1.078064 0.889793
H -0.435544 1.078064 -0.889793
C 0.048224 0.665550 0.000000
O 0.044194 -0.754448 0.000000
H -0.874480 -1.043721 0.000000
"""
mol.unit = "Angstrom"
mol.basis = "cc-pvtz"
mol.build()
<pyscf.gto.mole.Mole at 0x7aab94c83290>
mf = scf.RHF(mol).run()
mp2 = mp.MP2(mf).run()
converged SCF energy = -115.089057168863 E(MP2) = -115.546749317889 E_corr = -0.457692149025912 E(SCS-MP2) = -115.548492148244 E_corr = -0.459434979380392
opt = mp2.Gradients().optimizer(solver="geometric")
mol_eq = opt.kernel()
geometric-optimize called with the following command line:
/home/alex/miniconda3/envs/pyscf-dev/lib/python3.12/site-packages/ipykernel_launcher.py --f=/home/alex/.local/share/jupyter/runtime/kernel-v2-13676DEPx0bbU6O5e.json
())))))))))))))))/
())))))))))))))))))))))))),
*)))))))))))))))))))))))))))))))))
#, ()))))))))/ .)))))))))),
#%%%%, ()))))) .))))))))*
*%%%%%%, )) .. ,))))))).
*%%%%%%, ***************/. .)))))))
#%%/ (%%%%%%, /*********************. )))))))
.%%%%%%# *%%%%%%, *******/, **********, .))))))
.%%%%%%/ *%%%%%%, ** ******** .))))))
## .%%%%%%/ (%%%%%%, ,****** /)))))
%%%%%% .%%%%%%# *%%%%%%, ,/////. ****** ))))))
#% %% .%%%%%%/ *%%%%%%, ////////, *****/ ,)))))
#%% %%% %%%# .%%%%%%/ (%%%%%%, ///////. /***** ))))).
#%%%%. %%%%%# /%%%%%%* #%%%%%% /////) ****** ))))),
#%%%%##% %%%# .%%%%%%/ (%%%%%%, ///////. /***** ))))).
## %%% .%%%%%%/ *%%%%%%, ////////. *****/ ,)))))
#%%%%# /%%%%%%/ (%%%%%% /)/)// ****** ))))))
## .%%%%%%/ (%%%%%%, ******* ))))))
.%%%%%%/ *%%%%%%, **. /******* .))))))
*%%%%%%/ (%%%%%% ********/*..,*/********* *))))))
#%%/ (%%%%%%, *********************/ )))))))
*%%%%%%, ,**************/ ,))))))/
(%%%%%% () ))))))))
#%%%%, ()))))) ,)))))))),
#, ()))))))))) ,)))))))))).
()))))))))))))))))))))))))))))))/
())))))))))))))))))))))))).
())))))))))))))),
-=# geomeTRIC started. Version: 1.0 #=-
Current date and time: 2024-03-23 21:20:54
Custom engine selected.
Bonds will be generated from interatomic distances less than 1.20 times sum of covalent radii
18 internal coordinates being used (instead of 18 Cartesians)
Internal coordinate system (atoms numbered from 1):
Distance 1-4
Distance 2-4
Distance 3-4
Distance 4-5
Distance 5-6
Angle 1-4-2
Angle 1-4-3
Angle 1-4-5
Angle 2-4-3
Angle 2-4-5
Angle 3-4-5
Angle 4-5-6
Dihedral 1-4-5-6
Dihedral 2-4-5-6
Dihedral 3-4-5-6
Translation-X 1-6
Translation-Y 1-6
Translation-Z 1-6
Rotation-A 1-6
Rotation-B 1-6
Rotation-C 1-6
<class 'geometric.internal.Distance'> : 5
<class 'geometric.internal.Angle'> : 7
<class 'geometric.internal.Dihedral'> : 3
<class 'geometric.internal.TranslationX'> : 1
<class 'geometric.internal.TranslationY'> : 1
<class 'geometric.internal.TranslationZ'> : 1
<class 'geometric.internal.RotationA'> : 1
<class 'geometric.internal.RotationB'> : 1
<class 'geometric.internal.RotationC'> : 1
> ===== Optimization Info: ====
> Job type: Energy minimization
> Maximum number of optimization cycles: 300
> Initial / maximum trust radius (Angstrom): 0.100 / 0.300
> Convergence Criteria:
> Will converge when all 5 criteria are reached:
> |Delta-E| < 1.00e-06
> RMS-Grad < 3.00e-04
> Max-Grad < 4.50e-04
> RMS-Disp < 1.20e-03
> Max-Disp < 1.80e-03
> === End Optimization Info ===
Geometry optimization cycle 1
Cartesian coordinates (Angstrom)
Atom New coordinates dX dY dZ
H 1.090669 0.975611 0.000000 0.000000 0.000000 0.000000
H -0.435544 1.078064 0.889793 0.000000 0.000000 -0.000000
H -0.435544 1.078064 -0.889793 0.000000 0.000000 0.000000
C 0.048224 0.665550 0.000000 0.000000 0.000000 0.000000
O 0.044194 -0.754448 0.000000 0.000000 0.000000 0.000000
H -0.874480 -1.043721 0.000000 0.000000 0.000000 0.000000
converged SCF energy = -115.089057168865
E(MP2_Scanner) = -115.546749322394 E_corr = -0.457692153528722
E(SCS-MP2_Scanner) = -115.548492145681 E_corr = -0.459434976815418
--------------- MP2_Scanner gradients ---------------
x y z
0 H 0.0037113235 0.0009762788 0.0000000000
1 H -0.0019602440 0.0009230030 0.0032228600
2 H -0.0019602440 0.0009230030 -0.0032228600
3 C 0.0009918653 0.0009070122 -0.0000000000
4 O 0.0041423323 -0.0016285445 0.0000000000
5 H -0.0049250330 -0.0021007524 -0.0000000000
----------------------------------------------
cycle 1: E = -115.546749322 dE = -115.547 norm(grad) = 0.00975584
Step 0 : Gradient = 3.983e-03/5.354e-03 (rms/max) Energy = -115.5467493224 Hessian Eigenvalues: 2.30000e-02 5.00000e-02 5.00000e-02 ... 3.50939e-01 4.24598e-01 5.48032e-01
Geometry optimization cycle 2
Cartesian coordinates (Angstrom)
Atom New coordinates dX dY dZ
H 1.082552 0.971601 0.000000 -0.008117 -0.004010 0.000000
H -0.432970 1.076985 0.884536 0.002574 -0.001079 -0.005257
H -0.432970 1.076985 -0.884536 0.002574 -0.001079 0.005257
C 0.045743 0.662878 0.000000 -0.002481 -0.002672 0.000000
O 0.044989 -0.752475 0.000000 0.000795 0.001973 0.000000
H -0.869825 -1.036854 0.000000 0.004655 0.006867 0.000000
converged SCF energy = -115.089435766108
E(MP2_Scanner) = -115.546859964084 E_corr = -0.457424197976251
E(SCS-MP2_Scanner) = -115.548558979997 E_corr = -0.459123213889003
--------------- MP2_Scanner gradients ---------------
x y z
0 H -0.0001877465 -0.0004649158 0.0000000000
1 H 0.0000457271 -0.0000837216 -0.0001258365
2 H 0.0000457271 -0.0000837216 0.0001258365
3 C -0.0005118004 0.0008954399 -0.0000000000
4 O 0.0007734022 -0.0007348676 -0.0000000000
5 H -0.0001653095 0.0004717867 -0.0000000000
----------------------------------------------
cycle 2: E = -115.546859964 dE = -0.000110642 norm(grad) = 0.00165925
Step 1 : Displace = 6.317e-03/9.054e-03 (rms/max) Trust = 1.000e-01 (=) Grad = 6.774e-04/1.067e-03 (rms/max) E (change) = -115.5468599641 (-1.106e-04) Quality = 0.959 Hessian Eigenvalues: 2.30000e-02 5.00000e-02 5.00000e-02 ... 3.66299e-01 4.14907e-01 5.44078e-01
Geometry optimization cycle 3
Cartesian coordinates (Angstrom)
Atom New coordinates dX dY dZ
H 1.082897 0.973519 0.000000 0.000345 0.001918 -0.000000
H -0.432286 1.077334 0.884344 0.000684 0.000349 -0.000192
H -0.432286 1.077334 -0.884344 0.000684 0.000349 0.000192
C 0.046431 0.662731 0.000000 0.000688 -0.000147 -0.000000
O 0.043206 -0.752154 0.000000 -0.001783 0.000321 -0.000000
H -0.870442 -1.039645 0.000000 -0.000617 -0.002791 -0.000000
converged SCF energy = -115.08949678528
E(MP2_Scanner) = -115.546863613983 E_corr = -0.457366828702596
E(SCS-MP2_Scanner) = -115.548563758329 E_corr = -0.459066973048177
--------------- MP2_Scanner gradients ---------------
x y z
0 H -0.0001328572 0.0000353638 -0.0000000000
1 H 0.0000557015 -0.0001064424 -0.0001281383
2 H 0.0000557015 -0.0001064424 0.0001281383
3 C 0.0001528682 0.0005858007 0.0000000000
4 O -0.0001884534 -0.0003703000 -0.0000000000
5 H 0.0000570395 -0.0000379797 0.0000000000
----------------------------------------------
cycle 3: E = -115.546863614 dE = -3.6499e-06 norm(grad) = 0.000790234
Step 2 : Displace = 1.683e-03/2.861e-03 (rms/max) Trust = 1.414e-01 (+) Grad = 3.226e-04/6.054e-04 (rms/max) E (change) = -115.5468636140 (-3.650e-06) Quality = 0.916
Hessian Eigenvalues: 2.30000e-02 5.00000e-02 5.00000e-02 ... 3.49406e-01 4.33789e-01 5.43616e-01
Geometry optimization cycle 4
Cartesian coordinates (Angstrom)
Atom New coordinates dX dY dZ
H 1.082943 0.973054 0.000000 0.000046 -0.000465 0.000000
H -0.432390 1.077525 0.884539 -0.000104 0.000190 0.000195
H -0.432390 1.077525 -0.884539 -0.000104 0.000190 -0.000195
C 0.046194 0.662504 0.000000 -0.000237 -0.000227 0.000000
O 0.043388 -0.751885 0.000000 0.000182 0.000268 -0.000000
H -0.870226 -1.039603 -0.000000 0.000216 0.000042 -0.000000
converged SCF energy = -115.089498861653
E(MP2_Scanner) = -115.546864093532 E_corr = -0.45736523187897
E(SCS-MP2_Scanner) = -115.548562835516 E_corr = -0.459063973862505
--------------- MP2_Scanner gradients ---------------
x y z
0 H 0.0000030778 -0.0000065800 0.0000000000
1 H 0.0000037381 -0.0000278095 0.0000000211
2 H 0.0000037381 -0.0000278095 -0.0000000211
3 C 0.0000082656 0.0001563074 -0.0000000000
4 O -0.0000343972 -0.0000868525 0.0000000000
5 H 0.0000155777 -0.0000072558 -0.0000000000
----------------------------------------------
cycle 4: E = -115.546864094 dE = -4.79549e-07 norm(grad) = 0.000187482
Step 3 : Displace = 3.290e-04/4.659e-04 (rms/max) Trust = 2.000e-01 (+) Grad = 7.654e-05/1.565e-04 (rms/max) E (change) = -115.5468640935 (-4.795e-07) Quality = 1.218 Hessian Eigenvalues: 2.30000e-02 5.00000e-02 5.00000e-02 ... 3.49406e-01 4.33789e-01 5.43616e-01 Converged! =D #==========================================================================# #| If this code has benefited your research, please support us by citing: |# #| |# #| Wang, L.-P.; Song, C.C. (2016) "Geometry optimization made simple with |# #| translation and rotation coordinates", J. Chem, Phys. 144, 214108. |# #| http://dx.doi.org/10.1063/1.4952956 |# #==========================================================================# Time elapsed since start of run_optimizer: 24.833 seconds
xyz = mol_eq.atom_coords(unit="Angstrom")
atom_symbols = [mol_eq.atom_pure_symbol(i) for i in range(mol_eq.natm)]
xyz_str = f"{len(atom_symbols)}\n\n" + "".join(
[
f"{atom_symbols[i]} {xyz[i][0]:.12f} {xyz[i][1]:.12f} {xyz[i][2]:.12f}\n"
for i in range(len(atom_symbols))
]
)
print(xyz_str)
6 H 1.082942730243 0.973054295538 0.000000000000 H -0.432389937115 1.077524913433 0.884539060760 H -0.432389937115 1.077524913433 -0.884539060759 C 0.046194206046 0.662504379510 0.000000000000 O 0.043388186215 -0.751885455981 0.000000000000 H -0.870226248270 -1.039603045920 -0.000000000000
view = py3Dmol.view(width=400, height=400)
view.addModel(xyz_str, "xyz")
view.setStyle({"stick": {}})
view.zoomTo()
view.show()
3Dmol.js failed to load for some reason. Please check your browser console for error messages.
The final structure in Z-matrix would be
H
H 1 1.7577
H 1 1.7577 2 60.43
C 1 1.0823 2 36.02 3 323.22
O 4 1.4144 1 106.79 2 238.84
H 5 0.9578 4 107.59 1 180.00
Bond scans¶
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
from pyscf import gto, scf, mp, ci
import py3Dmol
HARTREE2KCALMOL = 627.5094737775374055927342256
def get_h2_string(r=0.72):
atom_string = f"""
H
H 1 {r}
"""
return atom_string
mol_h2 = gto.Mole()
mol_h2.atom = get_h2_string()
mol_h2.unit = "Angstrom"
mol_h2.basis = "aug-cc-pvtz"
mol_h2.charge = 0
mol_h2.spin = 0
mol_h2.verbose = 1
mol_h2.build()
<pyscf.gto.mole.Mole at 0x7cad682642c0>
r_h2_eq = 0.74
r_h2_scan = np.linspace(r_h2_eq - 0.4, r_h2_eq + 5, num=90)
r_h2_energies = np.empty(r_h2_scan.shape)
for i, r_h2 in enumerate(r_h2_scan):
mol_h2.atom = get_h2_string(r=r_h2)
mol_h2.build()
mf = scf.RHF(mol_h2)
mf.conv_tol = 1e-12
mf.run()
mycc = ci.CISD(mf)
mycc.run()
r_h2_energies[i] = mycc.e_tot
# Convert to kcal/mol and normalize from farthest
r_h2_energies *= HARTREE2KCALMOL
r_h2_energies -= r_h2_energies[-1]
plt.plot(r_h2_scan, r_h2_energies)
plt.xlabel("H-H distance [Å]")
plt.ylabel("Energy [kcal/mol]")
plt.axhline(y=0, color="dimgrey", linestyle=":")
df = pd.DataFrame({"r": r_h2_scan, "e": r_h2_energies})
df.to_csv("h2-scan.csv", index=False)

Methanol¶
def get_atom_string(
r_ch=1.0823, r_co=1.4144, r_oh=0.9578, a_och=106.79, a_coh=107.59, d_hcoh=180.00
):
atom_string = f"""
H
H 1 1.7577
H 1 1.7577 2 60.43
C 1 {r_ch} 2 36.02 3 323.22
O 4 {r_co} 1 {a_och} 2 238.84
H 5 {r_oh} 4 {a_coh} 1 {d_hcoh}
"""
return atom_string
mol = gto.Mole()
mol.atom = get_atom_string()
mol.unit = "Angstrom"
mol.basis = "cc-pvtz"
mol.verbose = 1
mol.build()
<pyscf.gto.mole.Mole at 0x7cad71e1ef90>
O-H bond¶
r_oh_eq = 0.9587
r_oh_scan = np.linspace(r_oh_eq - 0.2, r_oh_eq + 0.4, num=30)
r_oh_energies = np.empty(r_oh_scan.shape)
for i, r_oh in enumerate(r_oh_scan):
mol.atom = get_atom_string(r_oh=r_oh)
mol.build()
mf = scf.RHF(mol)
mf.conv_tol = 1e-12
mf.run()
mp2 = mp.MP2(mf)
mp2.conv_tol = 1e-12
mp2.run()
r_oh_energies[i] = mp2.e_tot_scs
# Convert to kcal/mol and normalize from farthest
r_oh_energies *= HARTREE2KCALMOL
r_oh_energies -= np.min(r_oh_energies)
df = pd.DataFrame({"r": r_oh_scan, "e": r_oh_energies})
df.to_csv("meoh-oh-scan.csv", index=False)

C-O¶
r_co_eq = 1.4144
r_co_scan = np.linspace(r_co_eq - 0.2, r_co_eq + 0.4, num=30)
r_co_energies = np.empty(r_co_scan.shape)
for i, r_co in enumerate(r_co_scan):
mol.atom = get_atom_string(r_co=r_co)
mol.build()
mf = scf.RHF(mol)
mf.conv_tol = 1e-12
mf.run()
mp2 = mp.MP2(mf)
mp2.conv_tol = 1e-12
mp2.run()
r_co_energies[i] = mp2.e_tot_scs
# Convert to kcal/mol and normalize from farthest
r_co_energies *= HARTREE2KCALMOL
r_co_energies -= np.min(r_co_energies)
df = pd.DataFrame({"r": r_co_scan, "e": r_co_energies})
df.to_csv("meoh-co-scan.csv", index=False)

C-H¶
r_ch_eq = 1.0823
r_ch_scan = np.linspace(r_ch_eq - 0.2, r_ch_eq + 0.4, num=30)
r_ch_energies = np.empty(r_ch_scan.shape)
for i, r_ch in enumerate(r_ch_scan):
mol.atom = get_atom_string(r_ch=r_ch)
mol.build()
mf = scf.RHF(mol)
mf.conv_tol = 1e-12
mf.run()
mp2 = mp.MP2(mf)
mp2.conv_tol = 1e-12
mp2.run()
r_ch_energies[i] = mp2.e_tot_scs
# Convert to kcal/mol and normalize from farthest
r_ch_energies *= HARTREE2KCALMOL
r_ch_energies -= np.min(r_ch_energies)
df = pd.DataFrame({"r": r_ch_scan, "e": r_ch_energies})
df.to_csv("meoh-ch-scan.csv", index=False)

plt.plot(r_oh_scan, r_oh_energies, color="#f94144", label="O-H")
plt.plot(r_co_scan, r_co_energies, color="dimgrey", label="C-O")
plt.plot(r_ch_scan, r_ch_energies, color="#277da1", label="C-H")
plt.xlabel("Distance [Å]")
plt.ylabel("Energy [kcal/mol]")
plt.axhline(y=0, color="dimgrey", linestyle=":")
plt.legend()
<matplotlib.legend.Legend at 0x7cad5f590560>

Bond orders¶
def get_ethane_string(r_cc=1.5214):
atom_string = f"""
H
H 1 1.7635
H 1 1.7635 2 60.00
C 1 1.0878 2 35.85 3 36.95
C 4 {r_cc} 1 110.61 2 121.37
H 5 1.0878 4 110.61 1 300.00
H 5 1.0878 4 110.61 1 60.00
H 5 1.0878 4 110.61 1 180.00
"""
return atom_string
def get_ethene_string(r_cc=1.3164):
atom_string = f"""
H
H 1 1.8280
C 2 1.0778 1 32.00
C 3 {r_cc} 2 122.00 1 180.00
H 4 1.0778 3 122.00 2 180.00
H 4 1.0778 3 122.00 2 0.00
"""
return atom_string
def get_ethyne_string(r_cc=1.1938):
atom_string = f"""
H
C 1 1.0497
C 2 {r_cc} 1 179.97
H 3 1.0497 2 179.97 1 179.09
"""
return atom_string
r_bond_order_scan = np.linspace(0.9, 1.9, num=20)
r_ethane_energies = np.empty(r_bond_order_scan.shape)
# Ethane
mol = gto.Mole()
mol.unit = "Angstrom"
mol.basis = "cc-pvtz"
mol.verbose = 1
for i, r in enumerate(r_bond_order_scan):
mol.atom = get_ethane_string(r_cc=r)
mol.build()
mf = scf.RHF(mol)
mf.conv_tol = 1e-12
mf.run()
mycc = ci.CISD(mf)
mycc.run()
r_ethane_energies[i] = mycc.e_tot
r_ethane_energies *= HARTREE2KCALMOL
r_ethane_energies -= np.min(r_ethane_energies)
df = pd.DataFrame({"r": r_bond_order_scan, "e": r_ethane_energies})
df.to_csv("ethane-scan.csv", index=False)
r_ethene_energies = np.empty(r_bond_order_scan.shape)
# ethene
mol = gto.Mole()
mol.unit = "Angstrom"
mol.basis = "cc-pvtz"
mol.verbose = 1
for i, r in enumerate(r_bond_order_scan):
mol.atom = get_ethene_string(r_cc=r)
mol.build()
mf = scf.RHF(mol)
mf.conv_tol = 1e-12
mf.run()
mycc = ci.CISD(mf)
mycc.run()
r_ethene_energies[i] = mycc.e_tot
r_ethene_energies *= HARTREE2KCALMOL
r_ethene_energies -= np.min(r_ethene_energies)
df = pd.DataFrame({"r": r_bond_order_scan, "e": r_ethene_energies})
df.to_csv("ethene-scan.csv", index=False)
r_ethyne_energies = np.empty(r_bond_order_scan.shape)
# ethyne
mol = gto.Mole()
mol.unit = "Angstrom"
mol.basis = "cc-pvtz"
mol.verbose = 1
for i, r in enumerate(r_bond_order_scan):
mol.atom = get_ethyne_string(r_cc=r)
mol.build()
mf = scf.RHF(mol)
mf.conv_tol = 1e-12
mf.run()
mycc = ci.CISD(mf)
mycc.run()
r_ethyne_energies[i] = mycc.e_tot
r_ethyne_energies *= HARTREE2KCALMOL
r_ethyne_energies -= np.min(r_ethyne_energies)
df = pd.DataFrame({"r": r_bond_order_scan, "e": r_ethyne_energies})
df.to_csv("ethyne-scan.csv", index=False)
plt.plot(r_bond_order_scan, r_ethane_energies, color="#01a0d7", label="Single")
plt.plot(r_bond_order_scan, r_ethene_energies, color="#025099", label="Double")
plt.plot(r_bond_order_scan, r_ethyne_energies, color="#03045e", label="Triple")
plt.xlabel("Distance [Å]")
plt.ylabel("Energy [kcal/mol]")
plt.axhline(y=0, color="dimgrey", linestyle=":")
plt.legend()
<matplotlib.legend.Legend at 0x7cad5f372d80>

Angle scans¶
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
from pyscf import gto, scf, ci
import py3Dmol
HARTREE2KCALMOL = 627.5094737775374055927342256
Water angle¶
def get_atom_string(r_oh1=0.9622, r_oh2=0.9622, a_hoh=104.63):
atom_string = f"""
O
H 1 {r_oh1}
H 1 {r_oh2} 2 {a_hoh}
"""
return atom_string
mol = gto.Mole()
mol.atom = get_atom_string()
mol.unit = "Angstrom"
mol.basis = "cc-pvtz"
mol.verbose = 1
mol.build()
<pyscf.gto.mole.Mole at 0x7e83f5e2ec00>
a_hoh_eq = 104.63
a_hoh_scan = np.linspace(70, 150, num=30)
a_hoh_energies = np.empty(a_hoh_scan.shape)
for i, a_hoh in enumerate(a_hoh_scan):
mol.atom = get_atom_string(a_hoh=a_hoh)
mol.build()
mf = scf.RHF(mol)
mf.conv_tol = 1e-12
mf.run()
mycc = ci.CISD(mf)
mycc.run()
a_hoh_energies[i] = mycc.e_tot
# Convert to kcal/mol and normalize from farthest
a_hoh_energies *= HARTREE2KCALMOL
a_hoh_energies -= np.min(a_hoh_energies)
df = pd.DataFrame({"r": a_hoh_scan, "e": a_hoh_energies})
df.to_csv("h2o-hoh-scan.csv", index=False)
plt.plot(a_hoh_scan, a_hoh_energies)
plt.xlabel("H-O-H angle [degrees]")
plt.ylabel("Energy [kcal/mol]")
plt.axhline(y=0, color="dimgrey", linestyle=":")
<matplotlib.lines.Line2D at 0x7e83f43ffd40>

Dihedral scans¶
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
from pyscf import gto, scf, mp
import py3Dmol
HARTREE2KCALMOL = 627.5094737775374055927342256
Ethane dihedral¶
def get_atom_string(d_hcch=180.00):
atom_string = f"""
H
H 1 1.7635
H 1 1.7635 2 60.00
C 1 1.0878 2 35.85 3 36.95
C 4 1.5214 1 110.61 2 121.37
H 5 1.0878 4 110.61 1 {d_hcch+120}
H 5 1.0878 4 110.61 1 {d_hcch-120}
H 5 1.0878 4 110.61 1 {d_hcch}
"""
return atom_string
mol = gto.Mole()
mol.atom = get_atom_string()
mol.unit = "Angstrom"
mol.basis = "cc-pvtz"
mol.verbose = 1
mol.build()
<pyscf.gto.mole.Mole at 0x7ceb457599d0>
d_hcch_scan = np.linspace(-180, 180, num=80)
d_hcch_energies = np.empty(d_hcch_scan.shape)
for i, d_hcch in enumerate(d_hcch_scan):
mol.atom = get_atom_string(d_hcch=d_hcch)
mol.build()
mf = scf.RHF(mol)
mf.run()
mp2 = mp.MP2(mf)
mp2.run()
d_hcch_energies[i] = mp2.e_tot_scs
d_hcch_energies *= HARTREE2KCALMOL
d_hcch_energies -= np.min(d_hcch_energies)
df = pd.DataFrame({"r": d_hcch_scan, "e": d_hcch_energies})
df.to_csv("ethane-hcch-scan.csv", index=False)
plt.plot(d_hcch_scan, d_hcch_energies)
plt.xlabel("H-C-C-H dihedral [degrees]")
plt.ylabel("Energy [kcal/mol]")
plt.xlim(-180, 180)
plt.axhline(y=0, color="dimgrey", linestyle=":")
<matplotlib.lines.Line2D at 0x7ceb3f9c69c0>

Ended: Quantum chemistry data
Force and energy
TODO:
We can obtain the energy, \(U\) due to this interaction by computing